

Gene expression distribution deconvolution in single-cell RNA sequencing
- PMID: 29946020
- PMCID: PMC6048536
- DOI: 10.1073/pnas.1721085115
Gene expression distribution deconvolution in single-cell RNA sequencing
Abstract
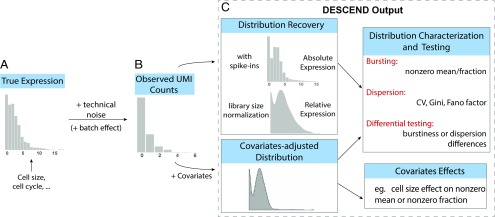
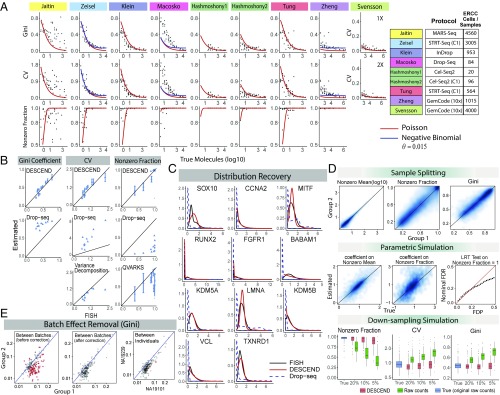
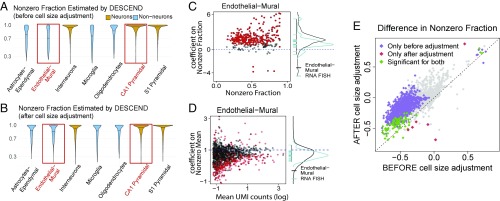
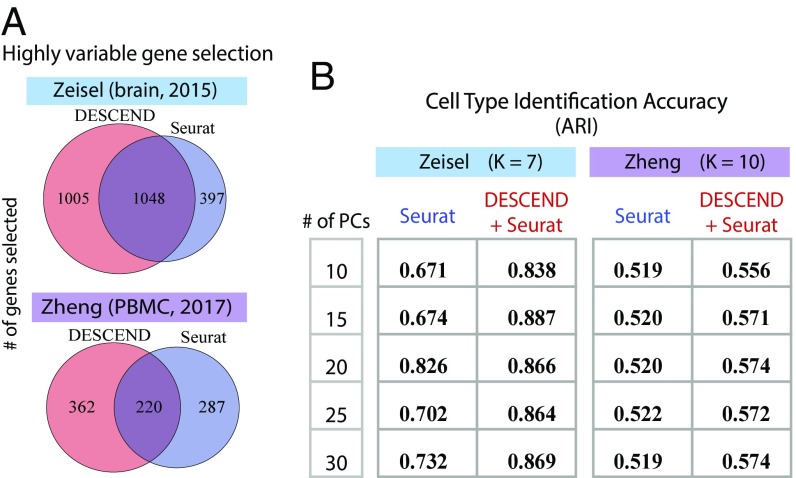
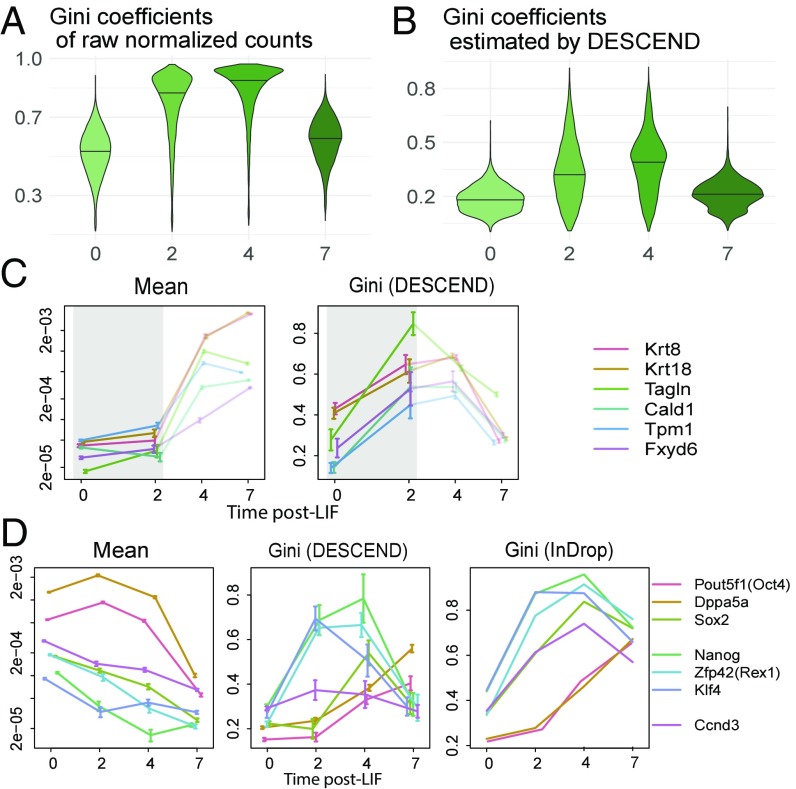
Single-cell RNA sequencing (scRNA-seq) enables the quantification of each gene's expression distribution across cells, thus allowing the assessment of the dispersion, nonzero fraction, and other aspects of its distribution beyond the mean. These statistical characterizations of the gene expression distribution are critical for understanding expression variation and for selecting marker genes for population heterogeneity. However, scRNA-seq data are noisy, with each cell typically sequenced at low coverage, thus making it difficult to infer properties of the gene expression distribution from raw counts. Based on a reexamination of nine public datasets, we propose a simple technical noise model for scRNA-seq data with unique molecular identifiers (UMI). We develop deconvolution of single-cell expression distribution (DESCEND), a method that deconvolves the true cross-cell gene expression distribution from observed scRNA-seq counts, leading to improved estimates of properties of the distribution such as dispersion and nonzero fraction. DESCEND can adjust for cell-level covariates such as cell size, cell cycle, and batch effects. DESCEND's noise model and estimation accuracy are further evaluated through comparisons to RNA FISH data, through data splitting and simulations and through its effectiveness in removing known batch effects. We demonstrate how DESCEND can clarify and improve downstream analyses such as finding differentially expressed genes, identifying cell types, and selecting differentiation markers.
Keywords: Gini coefficient; RNA sequencing; differential expression; highly variable genes; single-cell transcriptomics.
Copyright © 2018 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
