

Two-year progression analysis of RPE65 autosomal dominant retinitis pigmentosa
- PMID: 29947567
- PMCID: PMC6314189
- DOI: 10.1080/13816810.2018.1484929
Two-year progression analysis of RPE65 autosomal dominant retinitis pigmentosa
Abstract
Background: The RPE65 gene was recently described to cause autosomal dominant retinitis pigmentosa (adRP), presenting with a phenotype resembling choroideremia. This study presents the 2-year progression of RPE65 adRP in a patient.
Methods: This is an observational case report of one patient. The patient received a full ophthalmic examination during both visits, including diagnostic imaging such as spectral domain optical coherence tomography (SD-OCT), OCT-angiography (OCT-A), short-wave fundus autofluorescence (FAF), and fundus photography. Genetic characterization was obtained by DNA sequencing from peripheral blood lymphocytes obtained during the first visit.
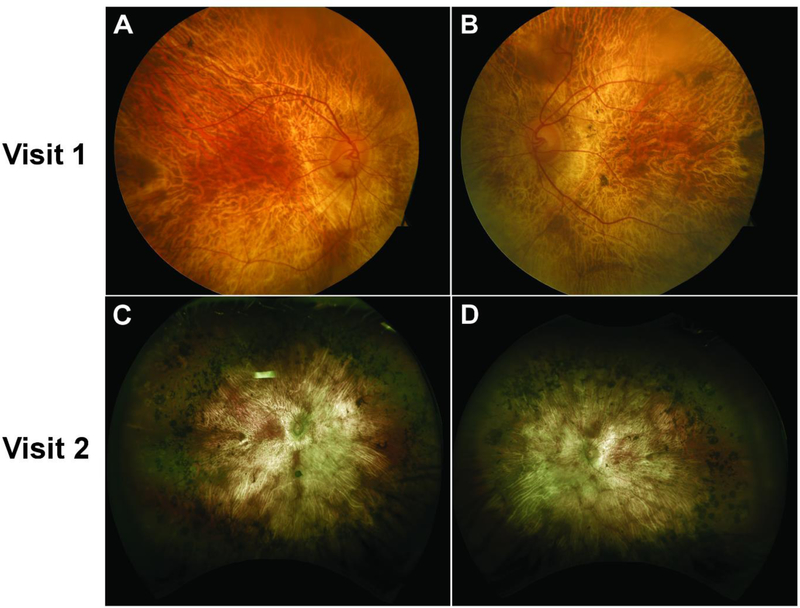
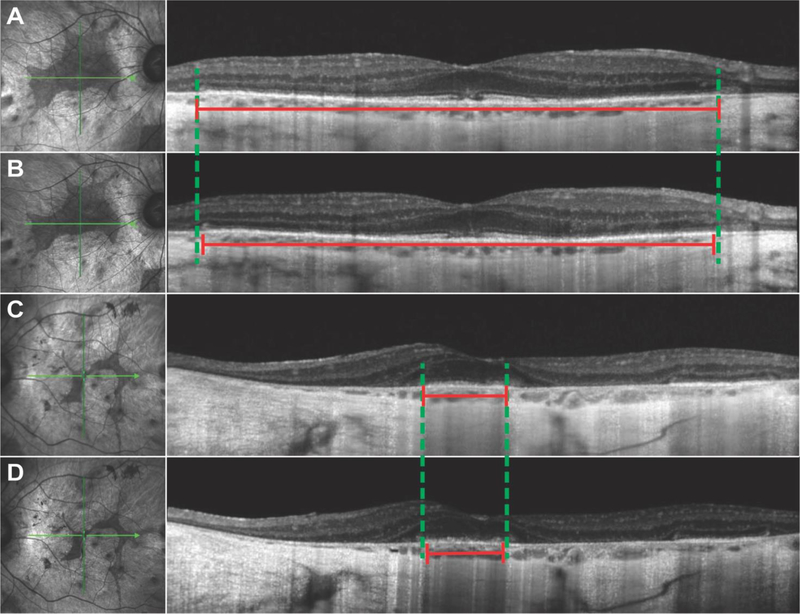
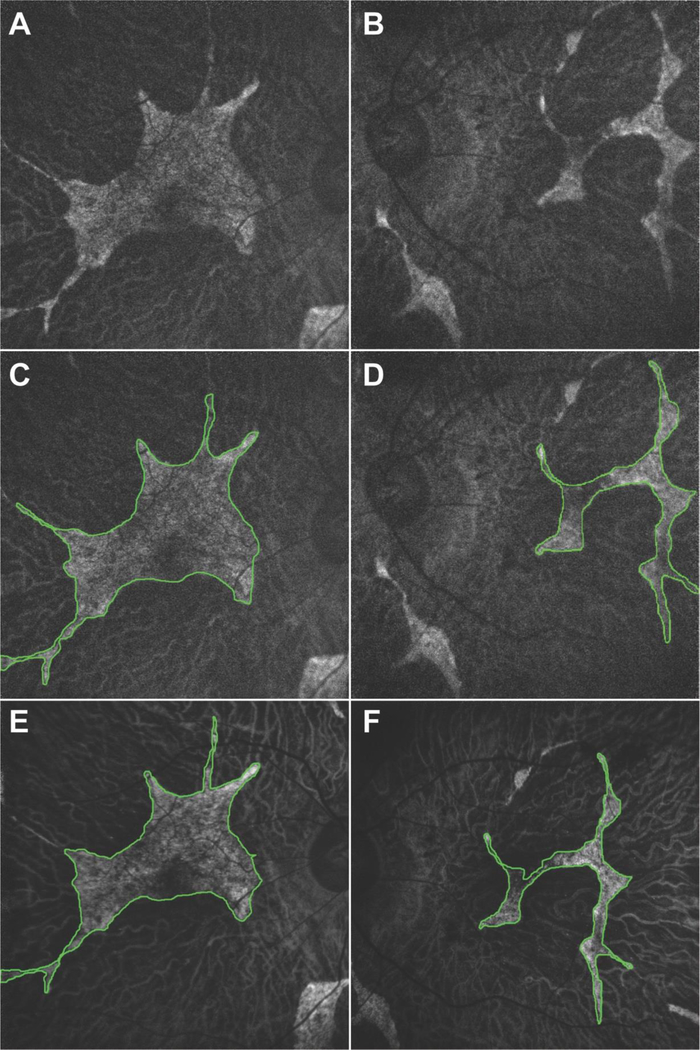
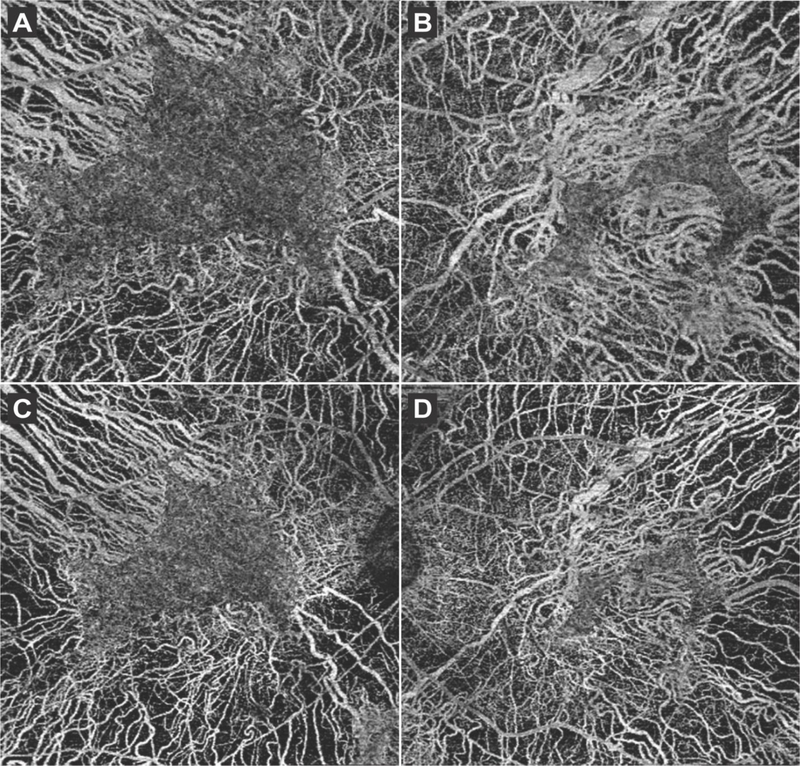
Results: RPE65 adRP phenocopied choroideremia at the initial fundoscopy. Upon the patient's return to our clinic 2 years later, DNA sequencing revealed a heterozygous mutation in the RPE65 gene. Diagnostic imaging by SD-OCT and FAF suggested disease progression. In conjunction with clinical examination and imaging, the diagnosis was revised to adRP caused by RPE65.
Conclusion: adRP due to a mutation in the gene encoding RPE65 phenocopied choroideremia. Based on our analysis of the 2-year disease progression in this patient, RPE65 adRP is mild and has a slow rate of disease progression.
Keywords: Autosomal dominant; RPE65; disease progression; retinitis pigmentosa.
Conflict of interest statement
DECLARATION OF INTEREST STATEMENT
The authors report no conflicts of interest.
Figures
References
-
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet 2006;368:1795–809. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials