

AmpUMI: design and analysis of unique molecular identifiers for deep amplicon sequencing
- PMID: 29949956
- PMCID: PMC6022702
- DOI: 10.1093/bioinformatics/bty264
AmpUMI: design and analysis of unique molecular identifiers for deep amplicon sequencing
Abstract
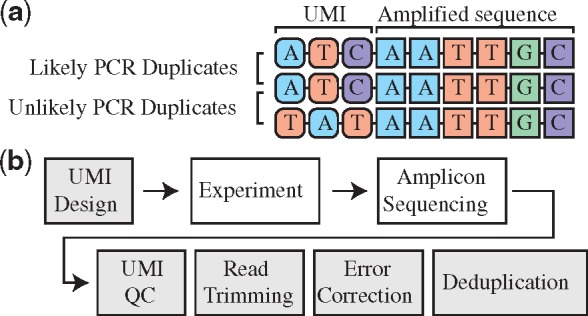
Motivation: Unique molecular identifiers (UMIs) are added to DNA fragments before PCR amplification to discriminate between alleles arising from the same genomic locus and sequencing reads produced by PCR amplification. While computational methods have been developed to take into account UMI information in genome-wide and single-cell sequencing studies, they are not designed for modern amplicon-based sequencing experiments, especially in cases of high allelic diversity. Importantly, no guidelines are provided for the design of optimal UMI length for amplicon-based sequencing experiments.
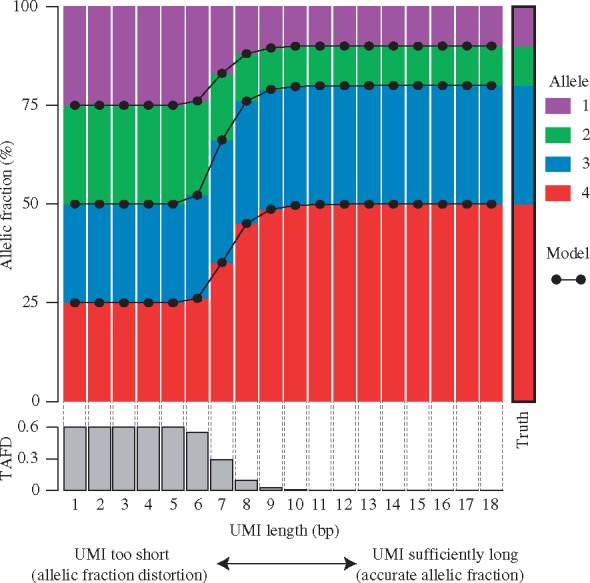
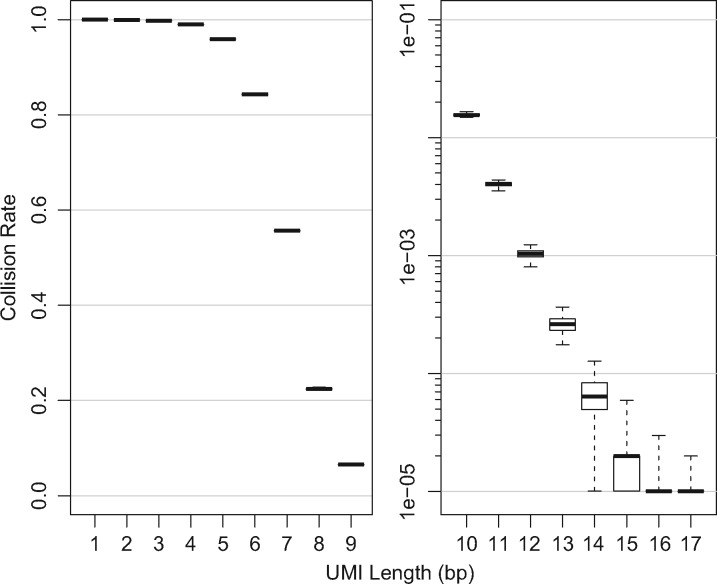
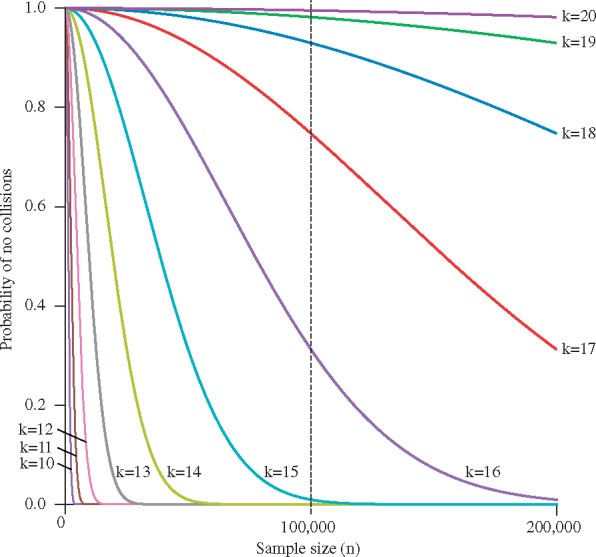
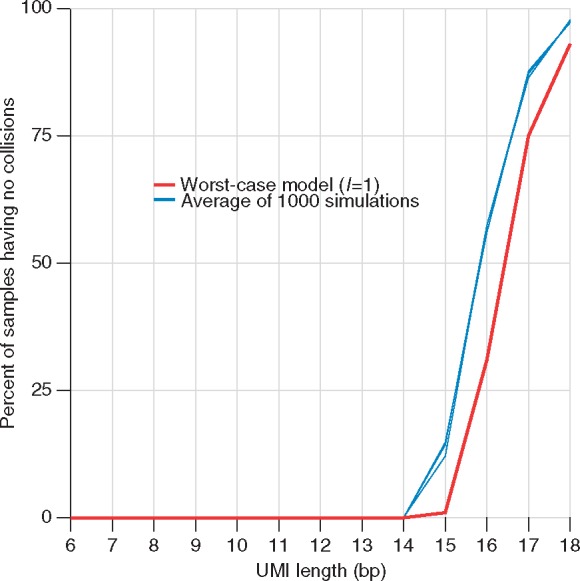
Results: Based on the total number of DNA fragments and the distribution of allele frequencies, we present a model for the determination of the minimum UMI length required to prevent UMI collisions and reduce allelic distortion. We also introduce a user-friendly software tool called AmpUMI to assist in the design and the analysis of UMI-based amplicon sequencing studies. AmpUMI provides quality control metrics on frequency and quality of UMIs, and trims and deduplicates amplicon sequences with user specified parameters for use in downstream analysis.
Availability and implementation: AmpUMI is open-source and freely available at http://github.com/pinellolab/AmpUMI.
Figures
References
-
- Islam S. et al. (2014) Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods, 11, 163–166. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
