

A graph-based approach to diploid genome assembly
- PMID: 29949989
- PMCID: PMC6022571
- DOI: 10.1093/bioinformatics/bty279
A graph-based approach to diploid genome assembly
Abstract
Motivation: Constructing high-quality haplotype-resolved de novo assemblies of diploid genomes is important for revealing the full extent of structural variation and its role in health and disease. Current assembly approaches often collapse the two sequences into one haploid consensus sequence and, therefore, fail to capture the diploid nature of the organism under study. Thus, building an assembler capable of producing accurate and complete diploid assemblies, while being resource-efficient with respect to sequencing costs, is a key challenge to be addressed by the bioinformatics community.
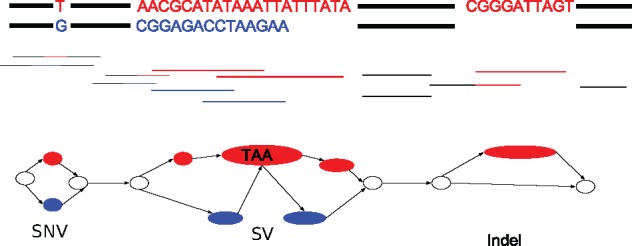
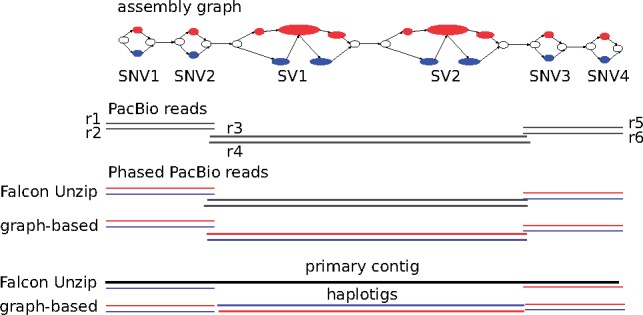
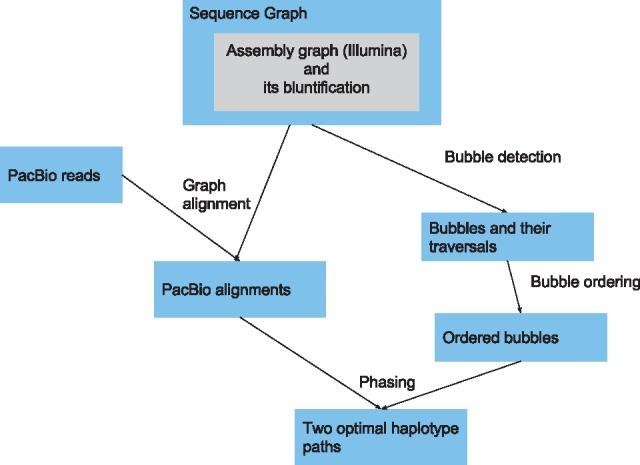
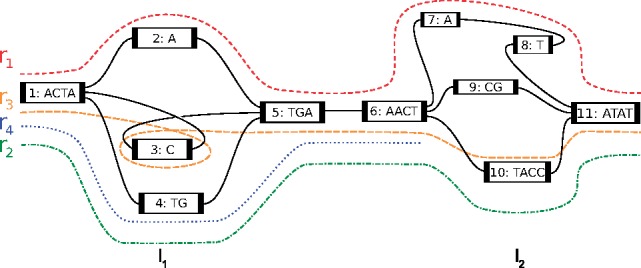
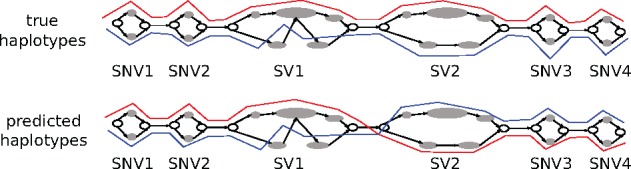
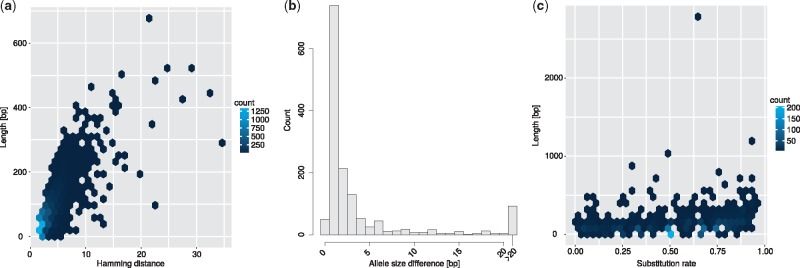
Results: We present a novel graph-based approach to diploid assembly, which combines accurate Illumina data and long-read Pacific Biosciences (PacBio) data. We demonstrate the effectiveness of our method on a pseudo-diploid yeast genome and show that we require as little as 50× coverage Illumina data and 10× PacBio data to generate accurate and complete assemblies. Additionally, we show that our approach has the ability to detect and phase structural variants.
Availability and implementation: https://github.com/whatshap/whatshap.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
References
-
- Berlin K. et al. (2015) Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol., 33, 623–630. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
