

HmmUFOtu: An HMM and phylogenetic placement based ultra-fast taxonomic assignment and OTU picking tool for microbiome amplicon sequencing studies
- PMID: 29950165
- PMCID: PMC6020470
- DOI: 10.1186/s13059-018-1450-0
HmmUFOtu: An HMM and phylogenetic placement based ultra-fast taxonomic assignment and OTU picking tool for microbiome amplicon sequencing studies
Abstract
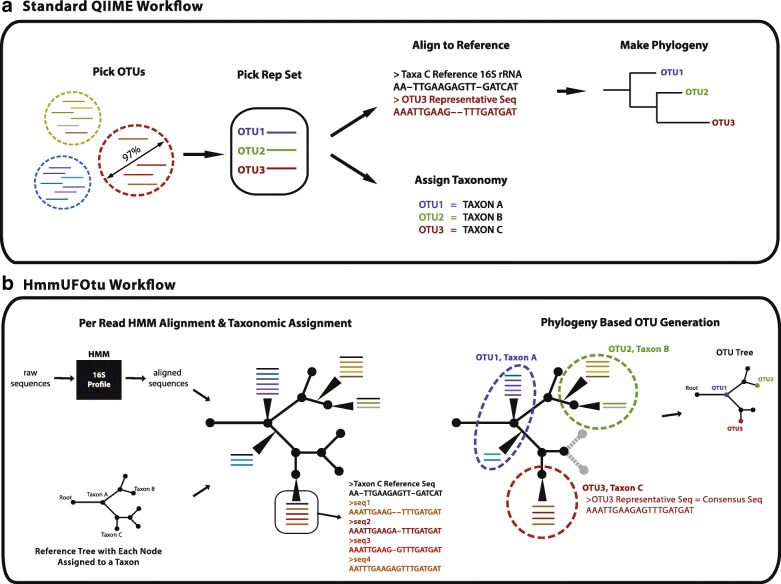
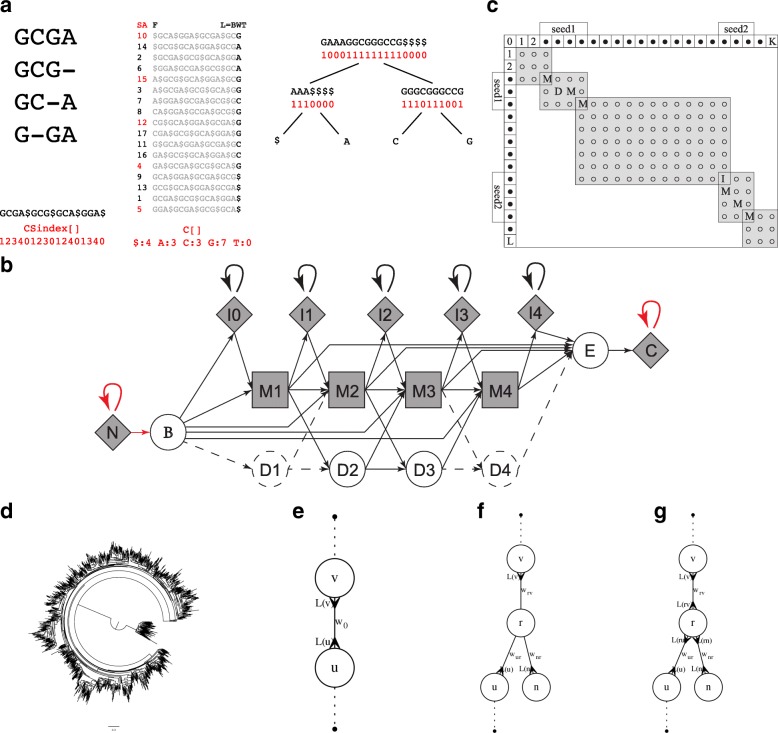
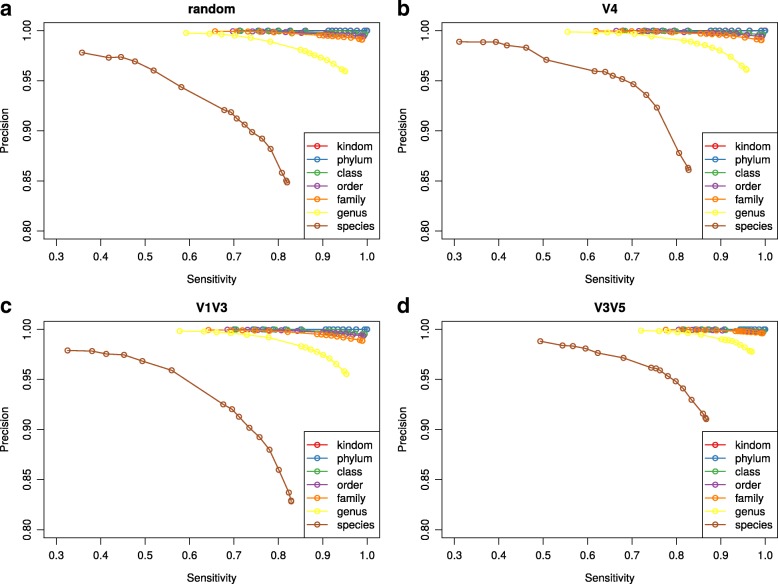
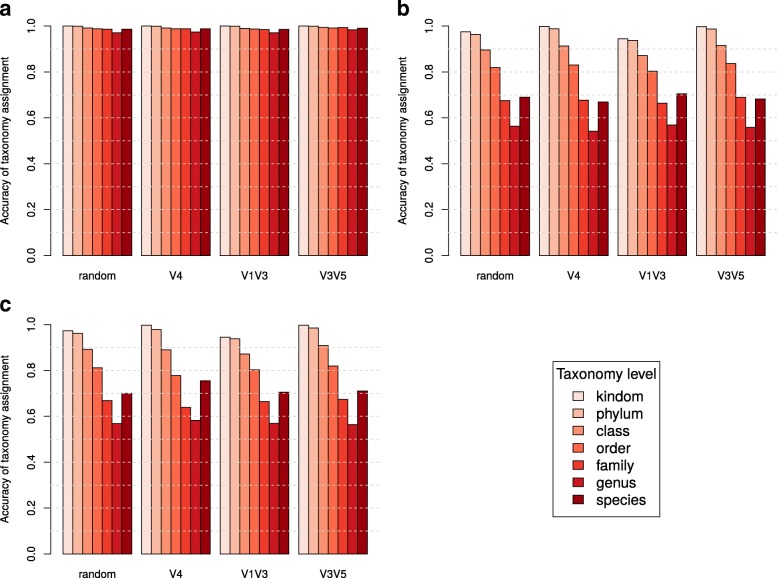
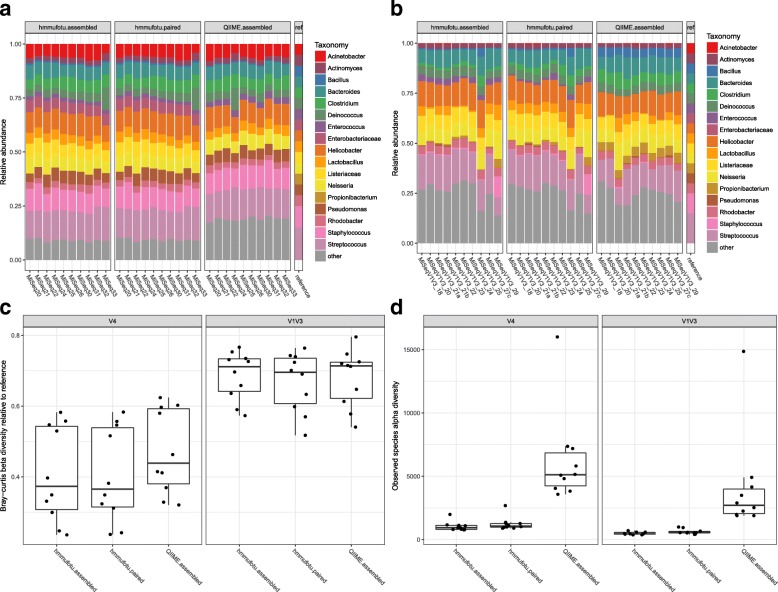
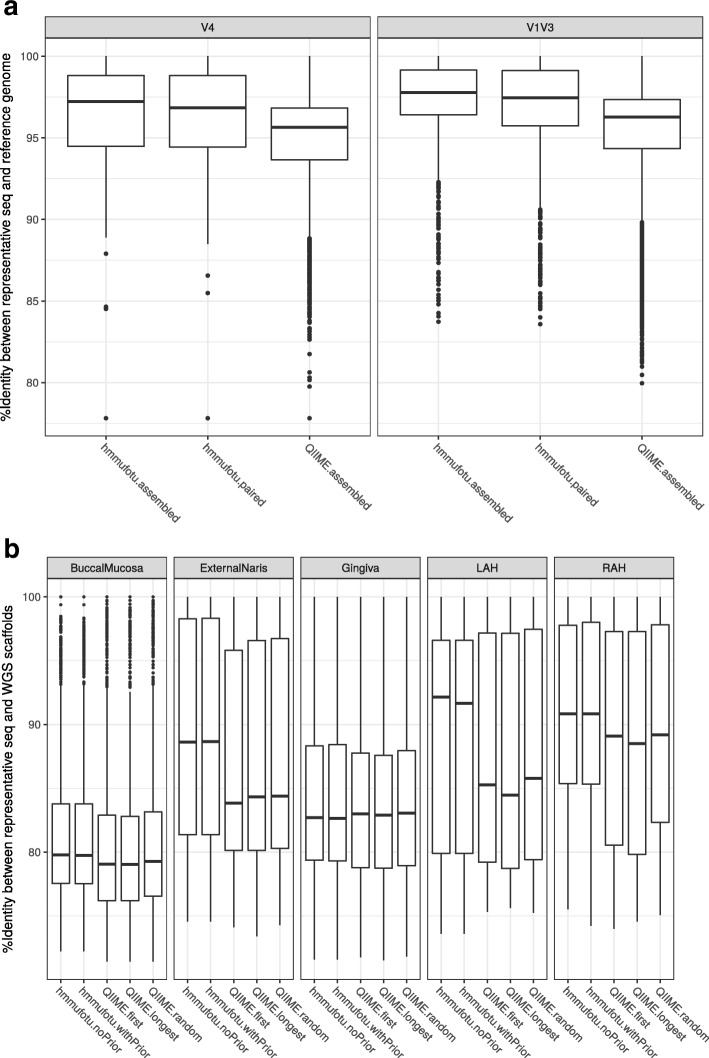
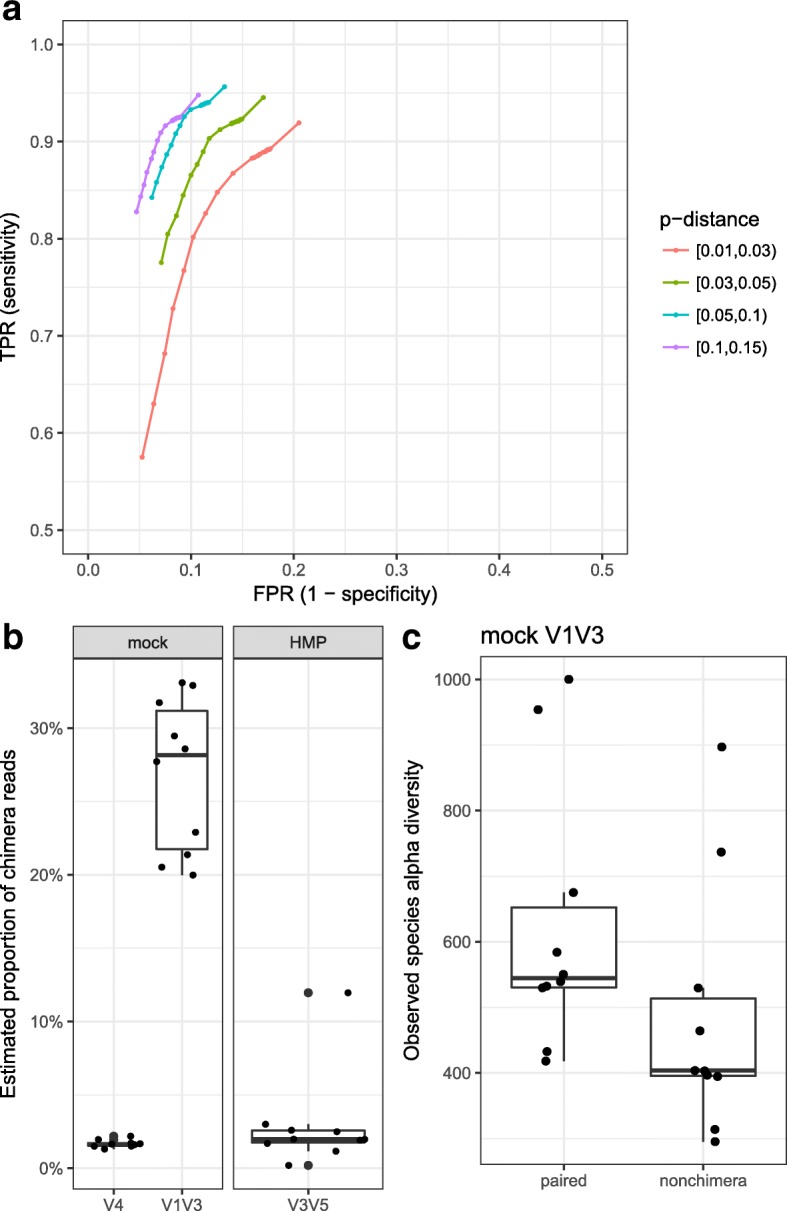
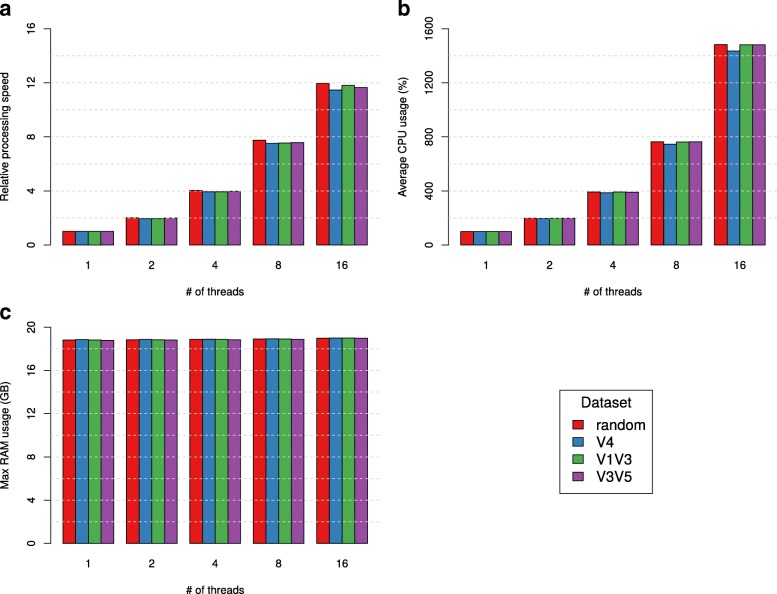
Culture-independent analysis of microbial communities frequently relies on amplification and sequencing of the prokaryotic 16S ribosomal RNA gene. Typical analysis pipelines group sequences into operational taxonomic units (OTUs) to infer taxonomic and phylogenetic relationships. Here, we present HmmUFOtu, a novel tool for processing microbiome amplicon sequencing data, which performs rapid per-read phylogenetic placement, followed by phylogenetically informed clustering into OTUs and taxonomy assignment. Compared to standard pipelines, HmmUFOtu more accurately and reliably recapitulates microbial community diversity and composition in simulated and real datasets without relying on heuristics or sacrificing speed or accuracy.
Keywords: 16S rRNA gene; DNA substitution models; Dirichlet models; FM-index; HMM profile alignment; Microbiome; Operational taxonomic unit; Phylogenetic placement; Taxonomic assignment.
Conflict of interest statement
Authors’ information
Jacquelyn S. Meisel, PhD is now a Postdoctoral Associate at the Institute of Advanced Computer Studies (UMIACS) at the University of Maryland College Park. The new contact email for Dr. Meisel is meiselj@umiacs.umd.edu.
Ethics approval and consent to participate
None of the authors have any ethics or consent issues to declare for the experimental data used in this study. HmmUFOtu is licensed under the GNU General Public License v3.0.
Consent for publication
All authors have consented to the publication of this work in
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01NR015639/NR/NINR NIH HHS/United States
- P30 AR069589/AR/NIAMS NIH HHS/United States
- R00AR060873/AR/NIAMS NIH HHS/United States
- R01 NR015639/NR/NINR NIH HHS/United States
- T32 AR007465/AR/NIAMS NIH HHS/United States
- T32AR007465/AR/NIAMS NIH HHS/United States
- R00 AR060873/AR/NIAMS NIH HHS/United States
- P30AR069589/AR/NIAMS NIH HHS/United States
- R01 AR066663/AR/NIAMS NIH HHS/United States
- T32HG00046/HG/NHGRI NIH HHS/United States
- R01AR066663/AR/NIAMS NIH HHS/United States
- T32 HG000046/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
