

Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line
- PMID: 29954844
- PMCID: PMC6071638
- DOI: 10.1101/gr.231100.117
Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line
Abstract
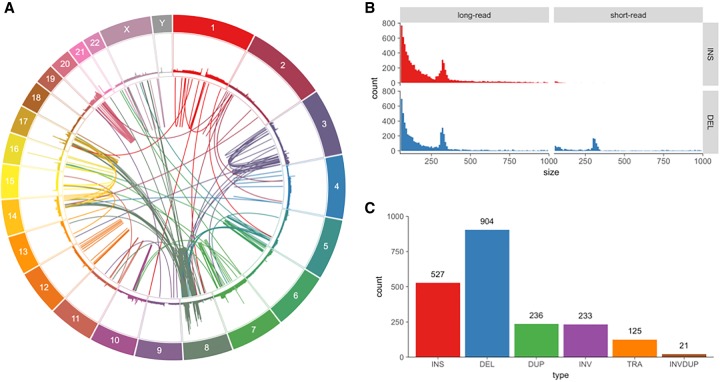
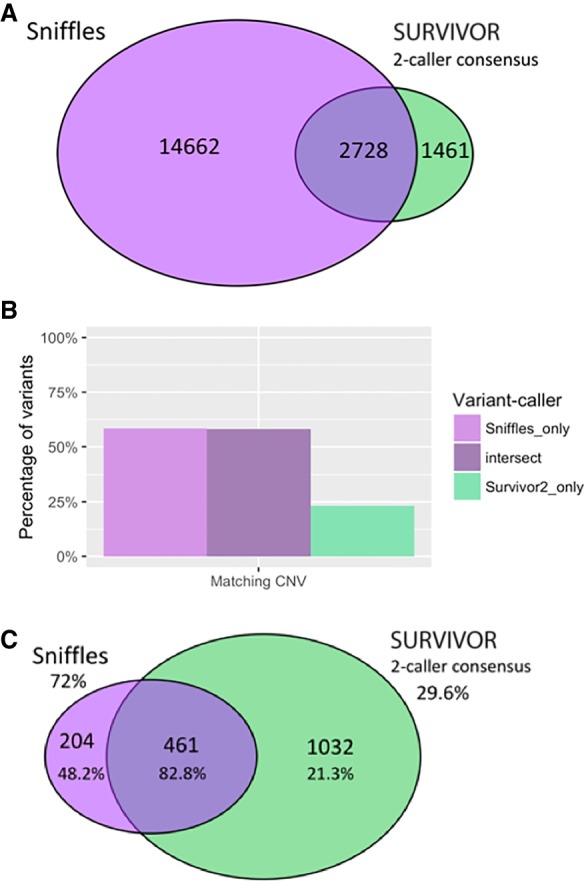
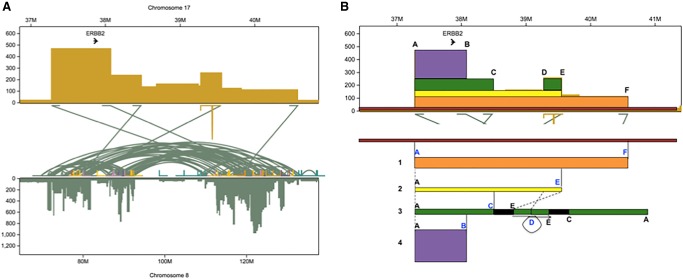
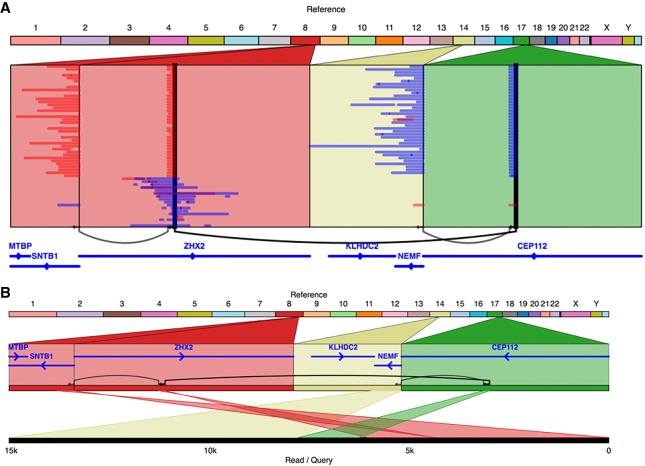
The SK-BR-3 cell line is one of the most important models for HER2+ breast cancers, which affect one in five breast cancer patients. SK-BR-3 is known to be highly rearranged, although much of the variation is in complex and repetitive regions that may be underreported. Addressing this, we sequenced SK-BR-3 using long-read single molecule sequencing from Pacific Biosciences and develop one of the most detailed maps of structural variations (SVs) in a cancer genome available, with nearly 20,000 variants present, most of which were missed by short-read sequencing. Surrounding the important ERBB2 oncogene (also known as HER2), we discover a complex sequence of nested duplications and translocations, suggesting a punctuated progression. Full-length transcriptome sequencing further revealed several novel gene fusions within the nested genomic variants. Combining long-read genome and transcriptome sequencing enables an in-depth analysis of how SVs disrupt the genome and sheds new light on the complex mechanisms involved in cancer genome evolution.
© 2018 Nattestad et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Arthur JG, Chen X, Zhou B, Urban AE, Wong WH. 2018. Detection of complex structural variation from paired-end sequencing data. bioRxiv 10.1101/200170. - DOI
-
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, Saunders CT. 2016. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32: 1220–1222. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous