

Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation
- PMID: 29955180
- PMCID: PMC6248311
- DOI: 10.1038/s41588-018-0154-4
Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation
Abstract
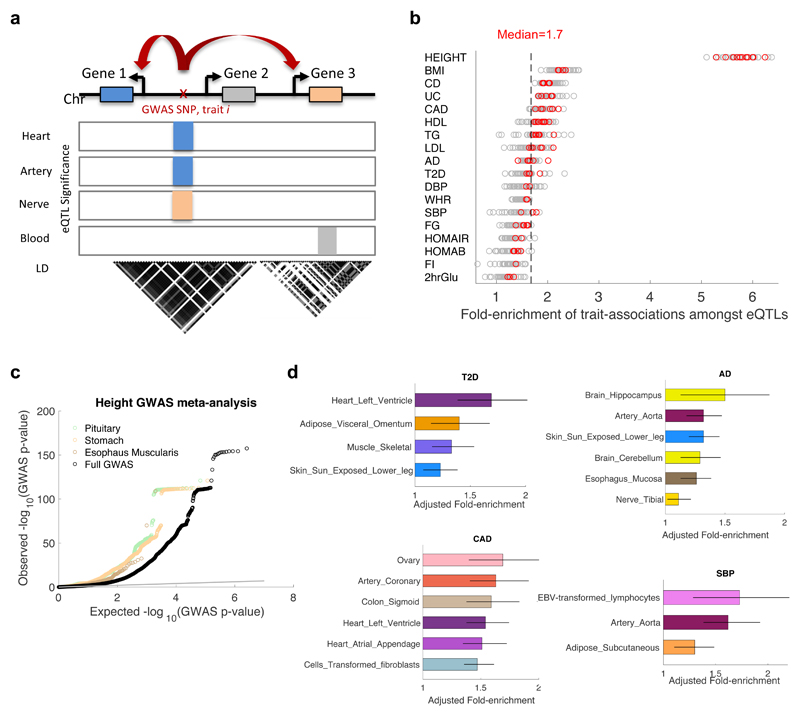
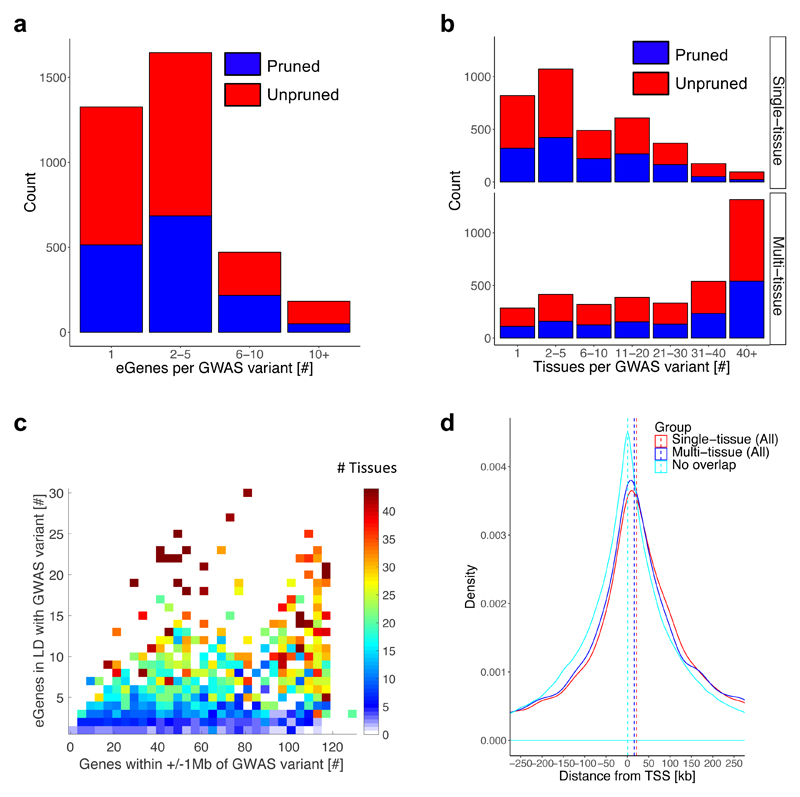
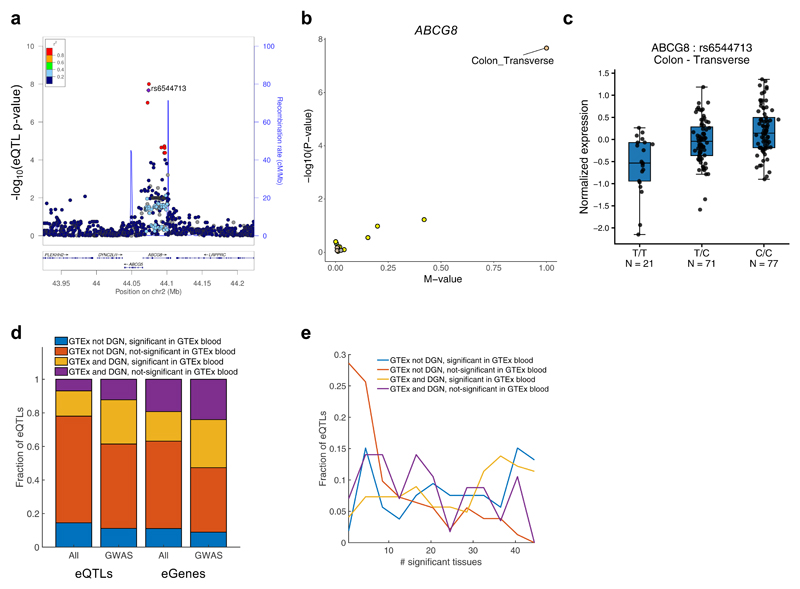
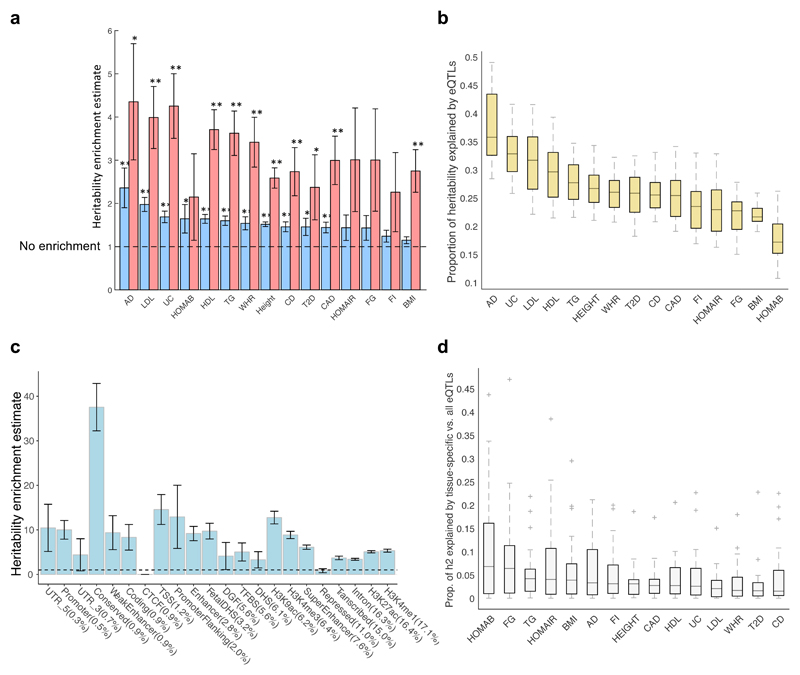
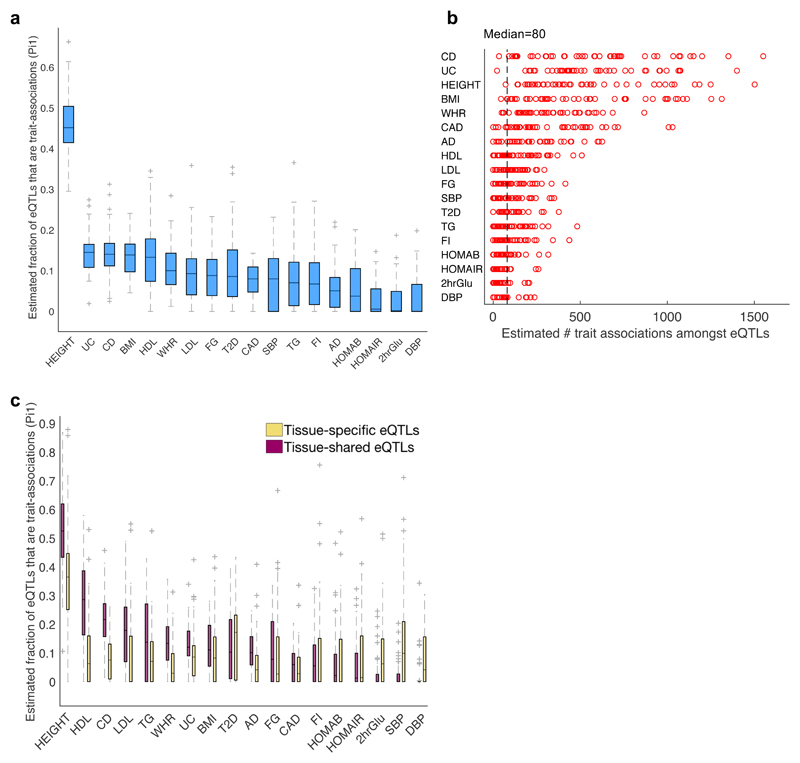
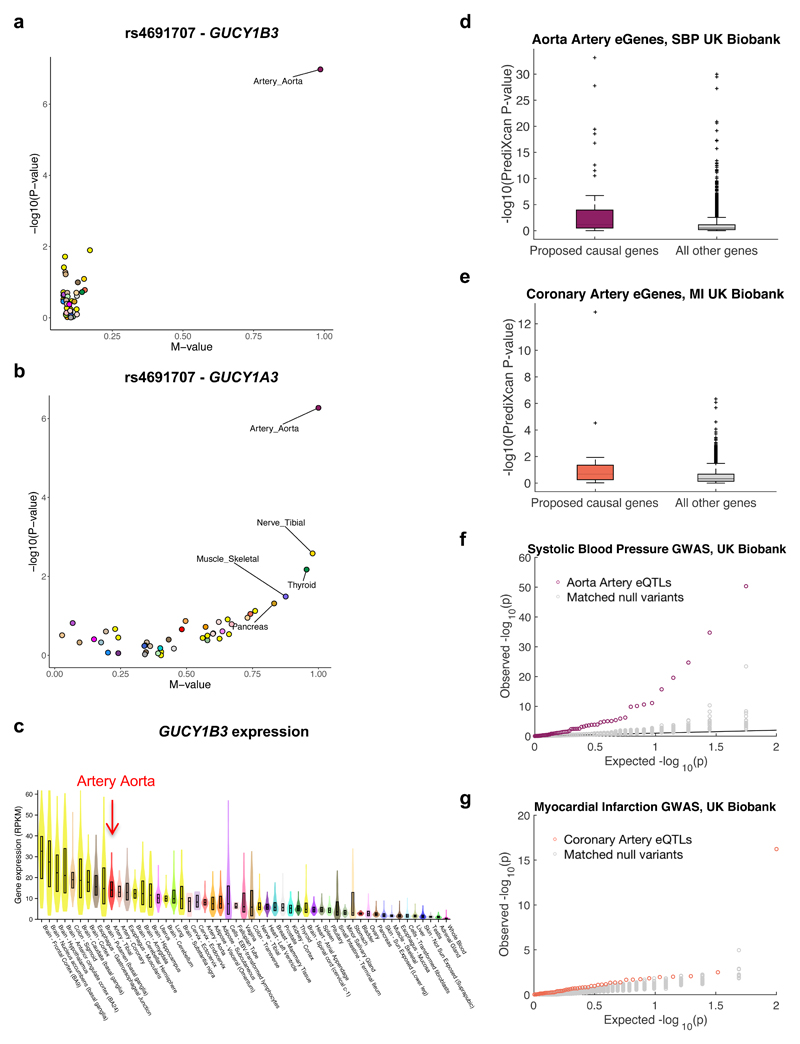
We apply integrative approaches to expression quantitative loci (eQTLs) from 44 tissues from the Genotype-Tissue Expression project and genome-wide association study data. About 60% of known trait-associated loci are in linkage disequilibrium with a cis-eQTL, over half of which were not found in previous large-scale whole blood studies. Applying polygenic analyses to metabolic, cardiovascular, anthropometric, autoimmune, and neurodegenerative traits, we find that eQTLs are significantly enriched for trait associations in relevant pathogenic tissues and explain a substantial proportion of the heritability (40-80%). For most traits, tissue-shared eQTLs underlie a greater proportion of trait associations, although tissue-specific eQTLs have a greater contribution to some traits, such as blood pressure. By integrating information from biological pathways with eQTL target genes and applying a gene-based approach, we validate previously implicated causal genes and pathways, and propose new variant and gene associations for several complex traits, which we replicate in the UK BioBank and BioVU.
Conflict of interest statement
M.I.McC serves on advisory panels for Pfizer and NovoNordisk. He has received honoraria from Pfizer, NovoNordisk, Sanofi-Aventis and Eli-Lilly, and research funding from Pfizer, Eli-Lilly, Merck, Takeda, Sanofi Aventis, Astra Zeneca, NovoNordisk, Servier, Janssen, Boehringer Ingelheim and Roche. M.v.d.B is an employee of Novo Nordisk. H.S.X. and J.Q. are employees of Pfizer.
Figures
References
Publication types
MeSH terms
Grants and funding
- UL1 TR000445/TR/NCATS NIH HHS/United States
- 203141/WT_/Wellcome Trust/United Kingdom
- U01 HG007610/HG/NHGRI NIH HHS/United States
- U19 HL065962/HL/NHLBI NIH HHS/United States
- S10 RR025141/RR/NCRR NIH HHS/United States
- 106130/WT_/Wellcome Trust/United Kingdom
- R01 HD074711/HD/NICHD NIH HHS/United States
- P50 GM115305/GM/NIGMS NIH HHS/United States
- R01 MH113362/MH/NIMH NIH HHS/United States
- R01 MH101814/MH/NIMH NIH HHS/United States
- MC_QA137853/MRC_/Medical Research Council/United Kingdom
- R01 MH101782/MH/NIMH NIH HHS/United States
- R01 AR042742/AR/NIAMS NIH HHS/United States
- R01 CA157823/CA/NCI NIH HHS/United States
- U01 DK105535/DK/NIDDK NIH HHS/United States
- 090532/WT_/Wellcome Trust/United Kingdom
- RC2 GM092618/GM/NIGMS NIH HHS/United States
- U01 HG006378/HG/NHGRI NIH HHS/United States
- R01 ES022282/ES/NIEHS NIH HHS/United States
- R01 MH101820/MH/NIMH NIH HHS/United States
- UL1 RR024975/RR/NCRR NIH HHS/United States
- R01 NS032830/NS/NINDS NIH HHS/United States
- U01 HG004798/HG/NHGRI NIH HHS/United States
- R01 HG007022/HG/NHGRI NIH HHS/United States
- MC_PC_17228/MRC_/Medical Research Council/United Kingdom
- U01 HG009088/HG/NHGRI NIH HHS/United States
- 098381/WT_/Wellcome Trust/United Kingdom
- R01 MH090937/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
