

Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species
- PMID: 29958539
- PMCID: PMC6025724
- DOI: 10.1186/s13072-018-0205-1
Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species
Abstract
Background: DNA methylation is one of the main epigenetic mechanisms for the regulation of gene expression in eukaryotes. In the standard model, methylation in gene promoters has received the most attention since it is generally associated with transcriptional silencing. Nevertheless, recent studies in human tissues reveal that methylation of the region downstream of the transcription start site is highly informative of gene expression. Also, in some cell types and specific genes it has been found that methylation of the first intron, a gene feature typically rich in enhancers, is linked with gene expression. However, a genome-wide, tissue-independent, systematic comparative analysis of the relationship between DNA methylation in the first intron and gene expression across vertebrates has not been explored yet.
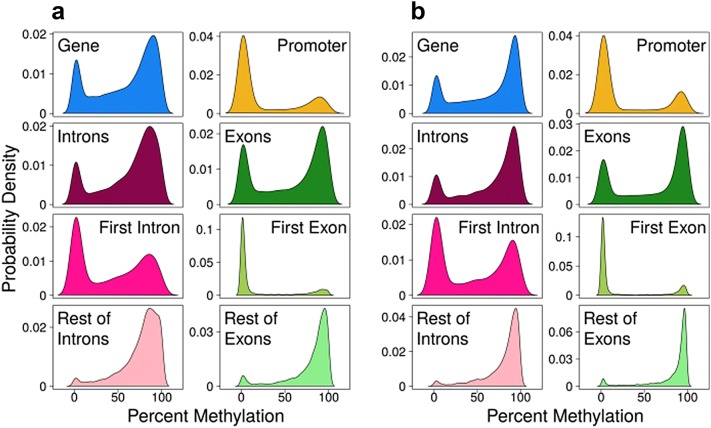
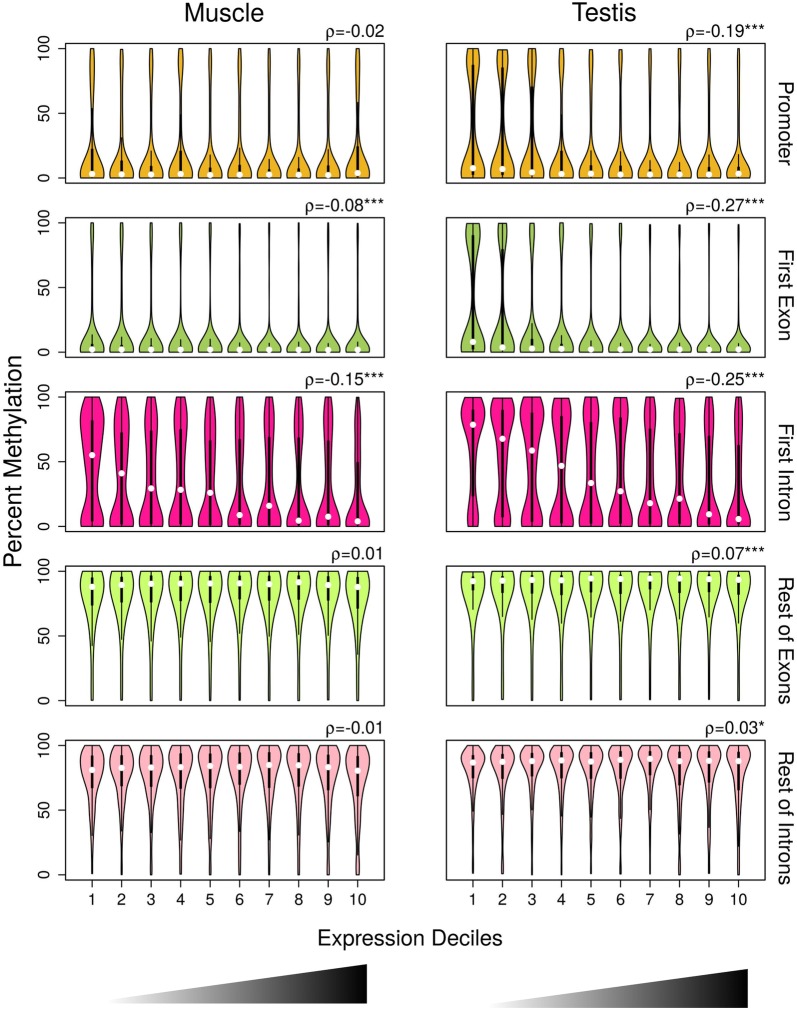
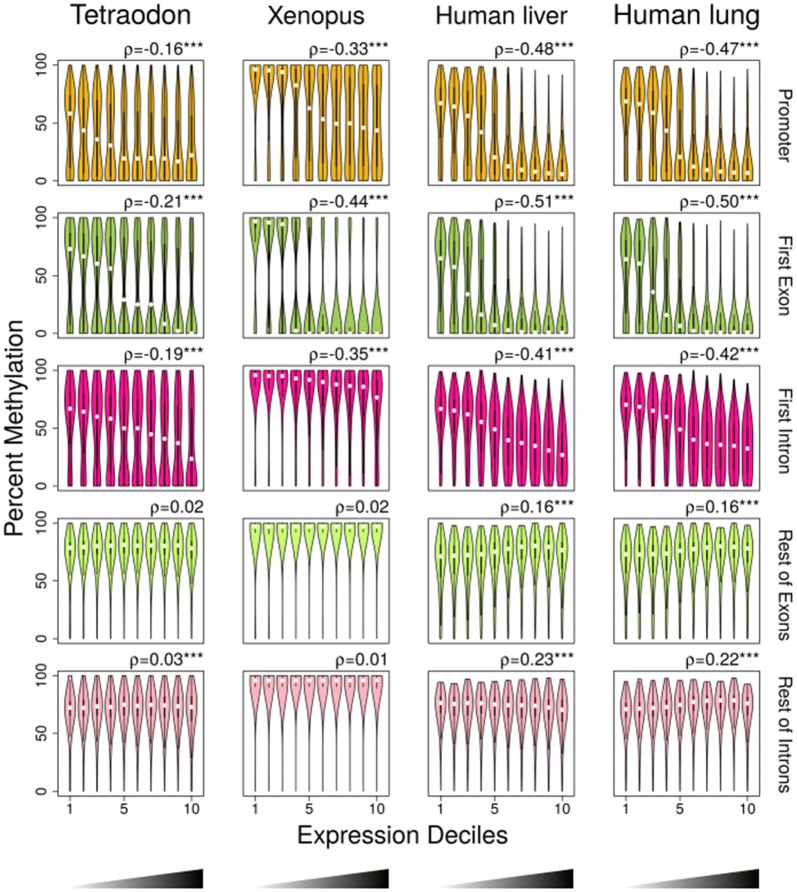
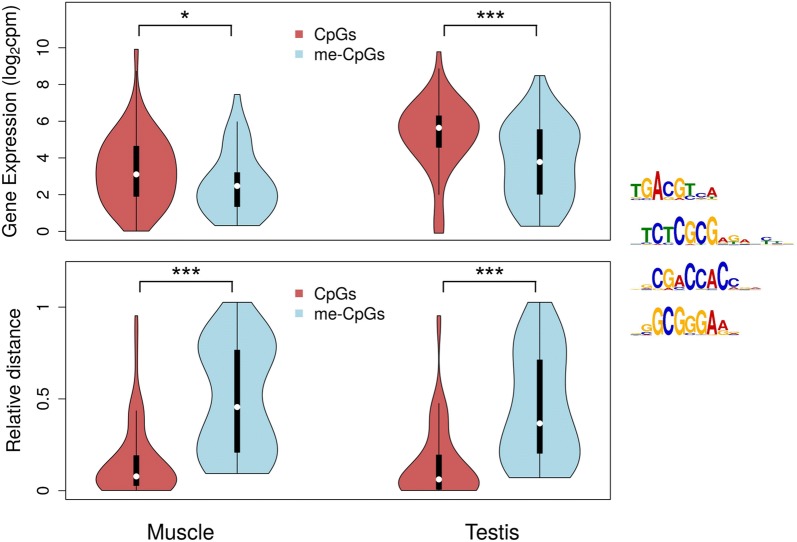
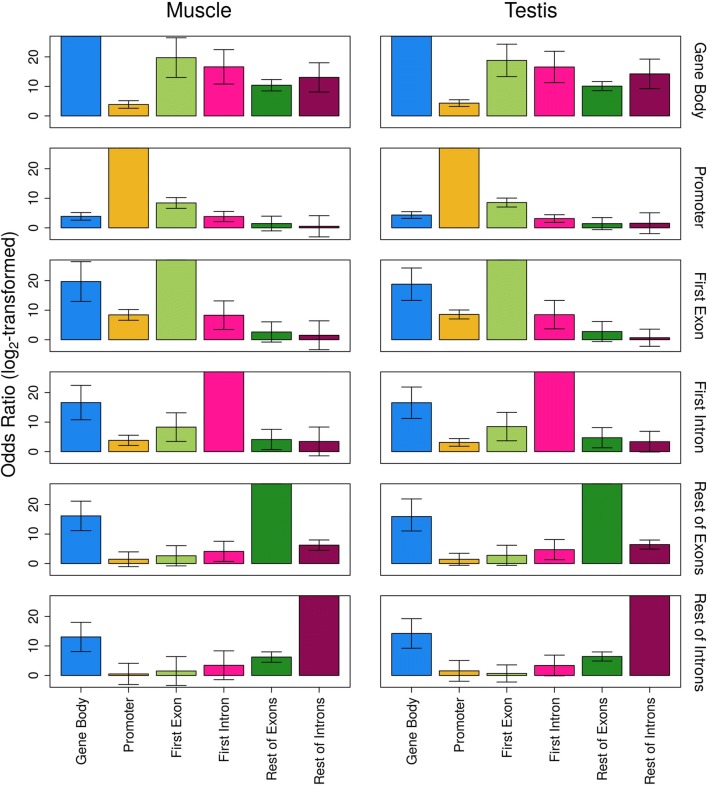
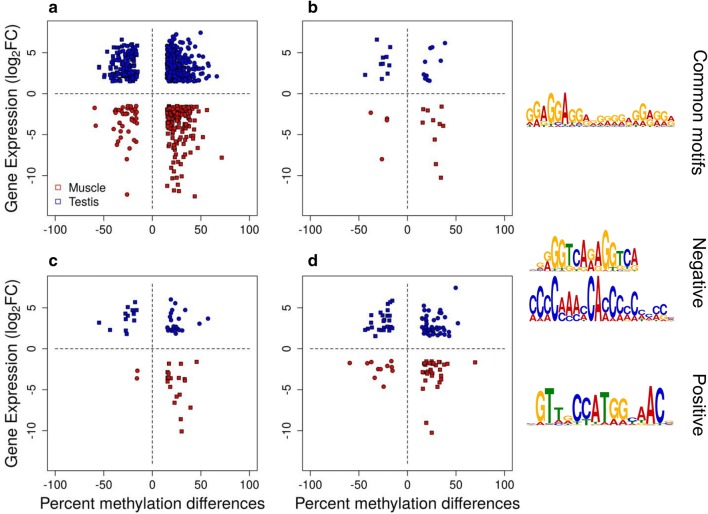
Results: The most important findings of this study are: (1) using different tissues from a modern fish, we show a clear genome-wide, tissue-independent quasi-linear inverse relationship between DNA methylation of the first intron and gene expression. (2) This relationship is conserved across vertebrates, since it is also present in the genomes of a model pufferfish, a model frog and different human tissues. Among the gene features, tissues and species interrogated, the first intron's negative correlation with the gene expression was most consistent. (3) We identified more tissue-specific differentially methylated regions (tDMRs) in the first intron than in any other gene feature. These tDMRs have positive or negative correlation with gene expression, indicative of distinct mechanisms of tissue-specific regulation. (4) Lastly, we identified CpGs in transcription factor binding motifs, enriched in the first intron, the methylation of which tended to increase with the distance from the first exon-first intron boundary, with a concomitant decrease in gene expression.
Conclusions: Our integrative analysis clearly reveals the important and conserved role of the methylation level of the first intron and its inverse association with gene expression regardless of tissue and species. These findings not only contribute to our basic understanding of the epigenetic regulation of gene expression but also identify the first intron as an informative gene feature regarding the relationship between DNA methylation and gene expression where future studies should be focused.
Keywords: DNA methylation; First intron; Gene expression; Gene features; Regulation.
Figures
References
-
- Gilbert SF, Epel D. Ecological developmental biology: integrating epigenetics, medicine, and evolution. Sunderland: Sinauer Associates; 2008.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
