

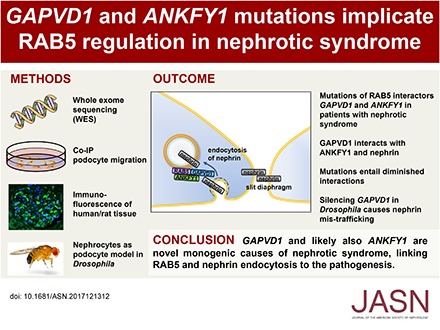
GAPVD1 and ANKFY1 Mutations Implicate RAB5 Regulation in Nephrotic Syndrome
- PMID: 29959197
- PMCID: PMC6065084
- DOI: 10.1681/ASN.2017121312
GAPVD1 and ANKFY1 Mutations Implicate RAB5 Regulation in Nephrotic Syndrome
Abstract
Background: Steroid-resistant nephrotic syndrome (SRNS) is a frequent cause of CKD. The discovery of monogenic causes of SRNS has revealed specific pathogenetic pathways, but these monogenic causes do not explain all cases of SRNS.
Methods: To identify novel monogenic causes of SRNS, we screened 665 patients by whole-exome sequencing. We then evaluated the in vitro functional significance of two genes and the mutations therein that we discovered through this sequencing and conducted complementary studies in podocyte-like Drosophila nephrocytes.
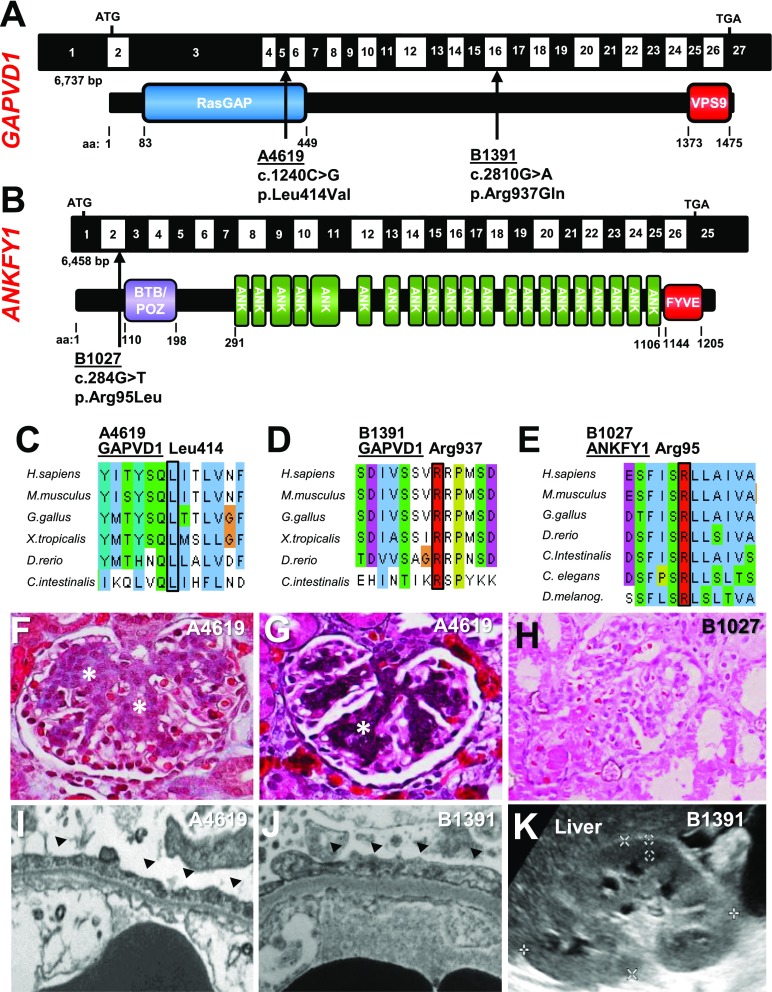
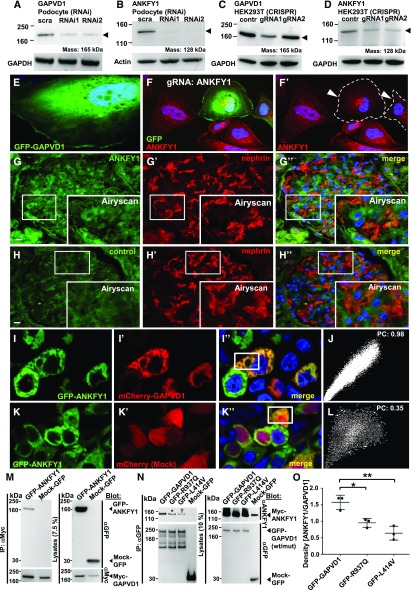
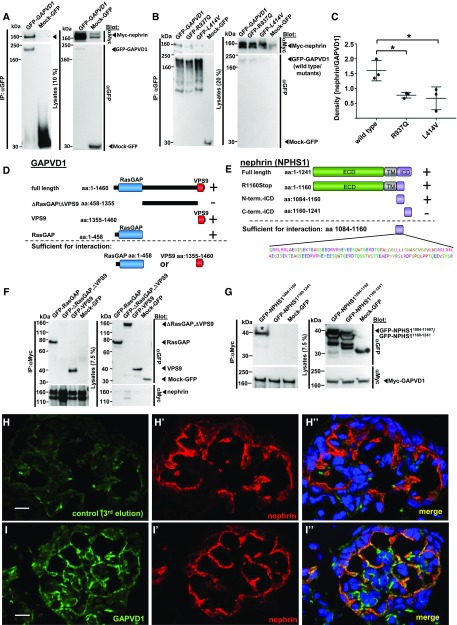
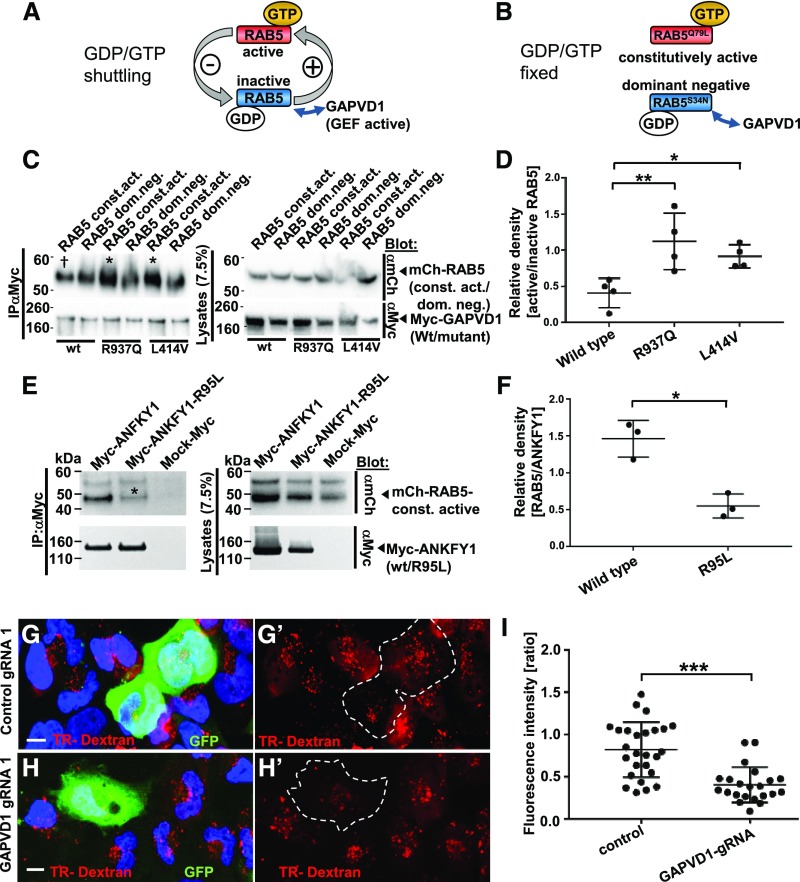
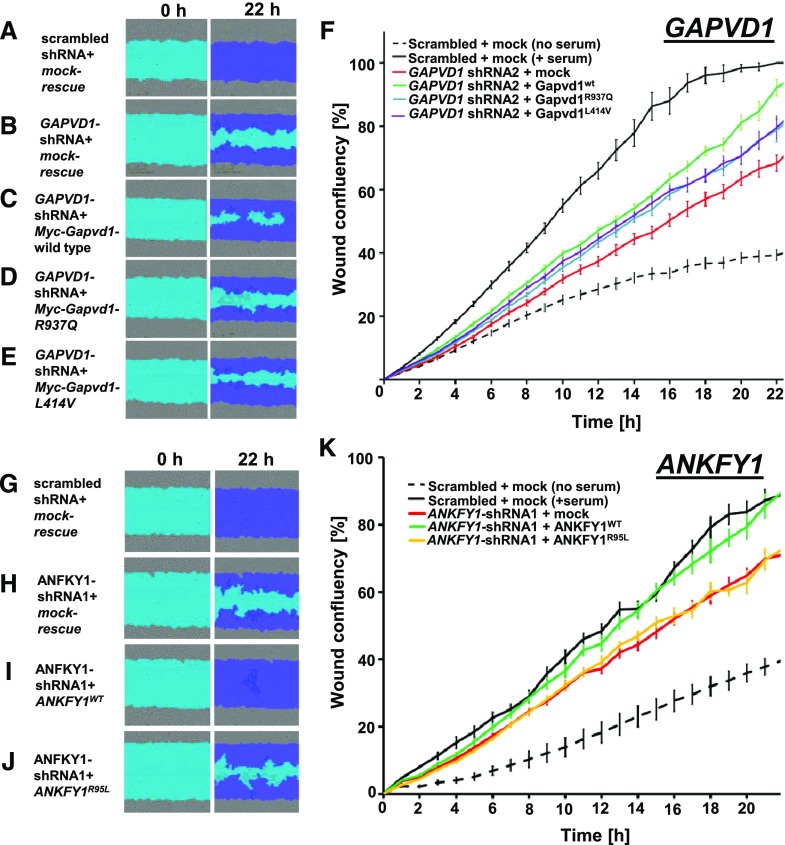
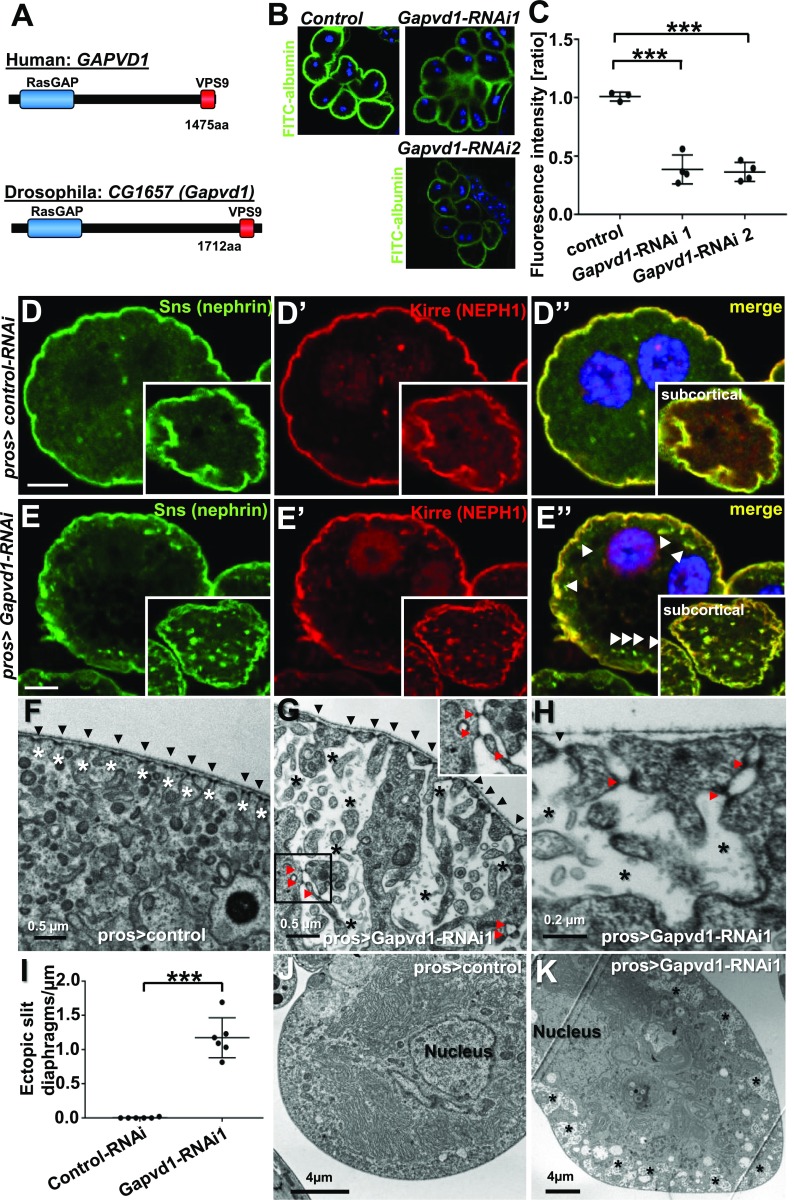
Results: We identified conserved, homozygous missense mutations of GAPVD1 in two families with early-onset NS and a homozygous missense mutation of ANKFY1 in two siblings with SRNS. GAPVD1 and ANKFY1 interact with the endosomal regulator RAB5. Coimmunoprecipitation assays indicated interaction between GAPVD1 and ANKFY1 proteins, which also colocalized when expressed in HEK293T cells. Silencing either protein diminished the podocyte migration rate. Compared with wild-type GAPVD1 and ANKFY1, the mutated proteins produced upon ectopic expression of GAPVD1 or ANKFY1 bearing the patient-derived mutations exhibited altered binding affinity for active RAB5 and reduced ability to rescue the knockout-induced defect in podocyte migration. Coimmunoprecipitation assays further demonstrated a physical interaction between nephrin and GAPVD1, and immunofluorescence revealed partial colocalization of these proteins in rat glomeruli. The patient-derived GAPVD1 mutations reduced nephrin-GAPVD1 binding affinity. In Drosophila, silencing Gapvd1 impaired endocytosis and caused mistrafficking of the nephrin ortholog.
Conclusions: Mutations in GAPVD1 and probably in ANKFY1 are novel monogenic causes of NS. The discovery of these genes implicates RAB5 regulation in the pathogenesis of human NS.
Keywords: endocytosis; genetic renal disease; nephrin; nephrocyte; nephrotic syndrome; podocyte.
Copyright © 2018 by the American Society of Nephrology.
Figures
References
-
- Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, et al.: Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol Cell 1: 575–582, 1998 - PubMed
-
- Löwik MM, Groenen PJ, Pronk I, Lilien MR, Goldschmeding R, Dijkman HB, et al.: Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney Int 72: 1198–1203, 2007 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
