

Detection of Splicing Abnormalities and Genotype-Phenotype Correlation in X-linked Alport Syndrome
- PMID: 29959198
- PMCID: PMC6065097
- DOI: 10.1681/ASN.2018030228
Detection of Splicing Abnormalities and Genotype-Phenotype Correlation in X-linked Alport Syndrome
Abstract
Background: X-linked Alport syndrome (XLAS) is a progressive hereditary nephropathy caused by mutations in the COL4A5 gene. Genotype-phenotype correlation in male XLAS is relatively well established; relative to truncating mutations, nontruncating mutations exhibit milder phenotypes. However, transcript comparison between XLAS cases with splicing abnormalities that result in a premature stop codon and those with nontruncating splicing abnormalities has not been reported, mainly because transcript analysis is not routinely conducted in patients with XLAS.
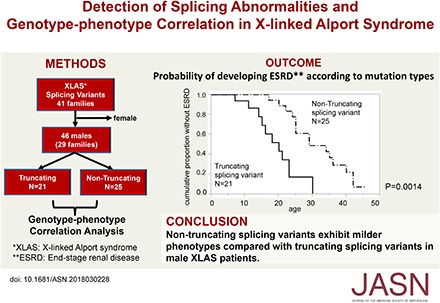
Methods: We examined transcript expression for all patients with suspected splicing abnormalities who were treated at one hospital between January of 2006 and July of 2017. Additionally, we recruited 46 males from 29 families with splicing abnormalities to examine genotype-phenotype correlation in patients with truncating (n=21, from 14 families) and nontruncating (n=25, from 15 families) mutations at the transcript level.
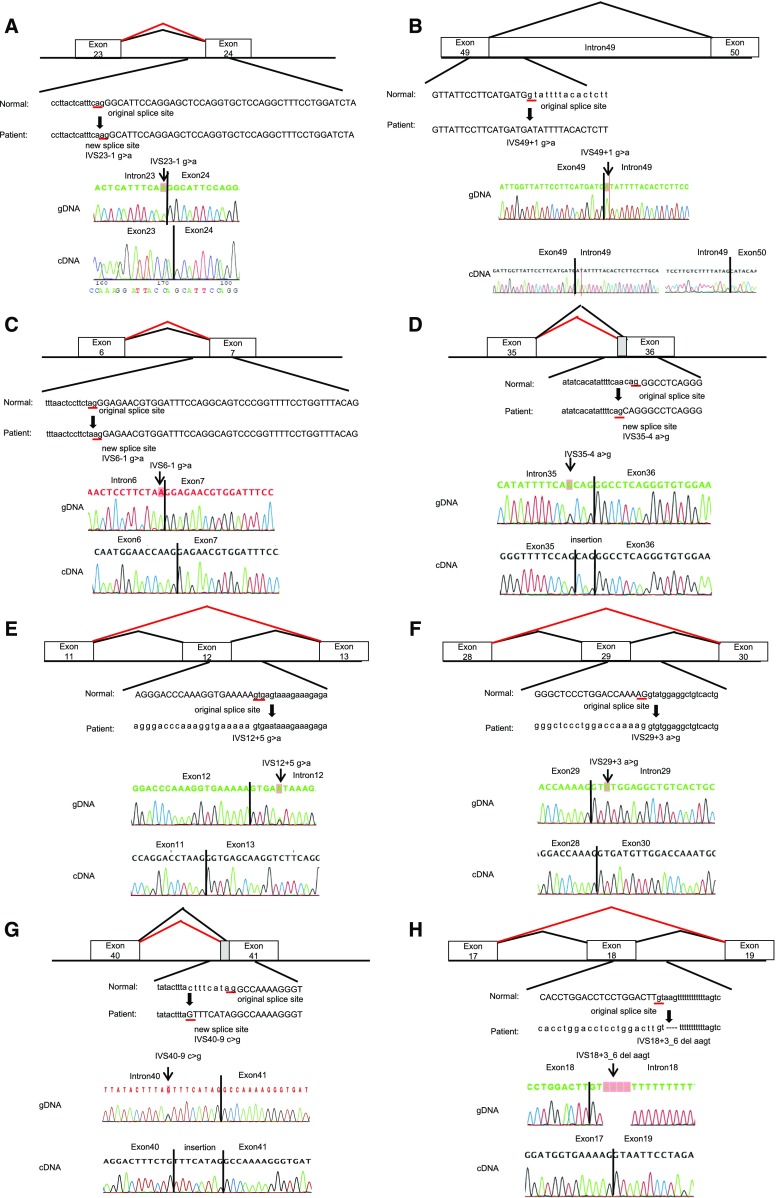
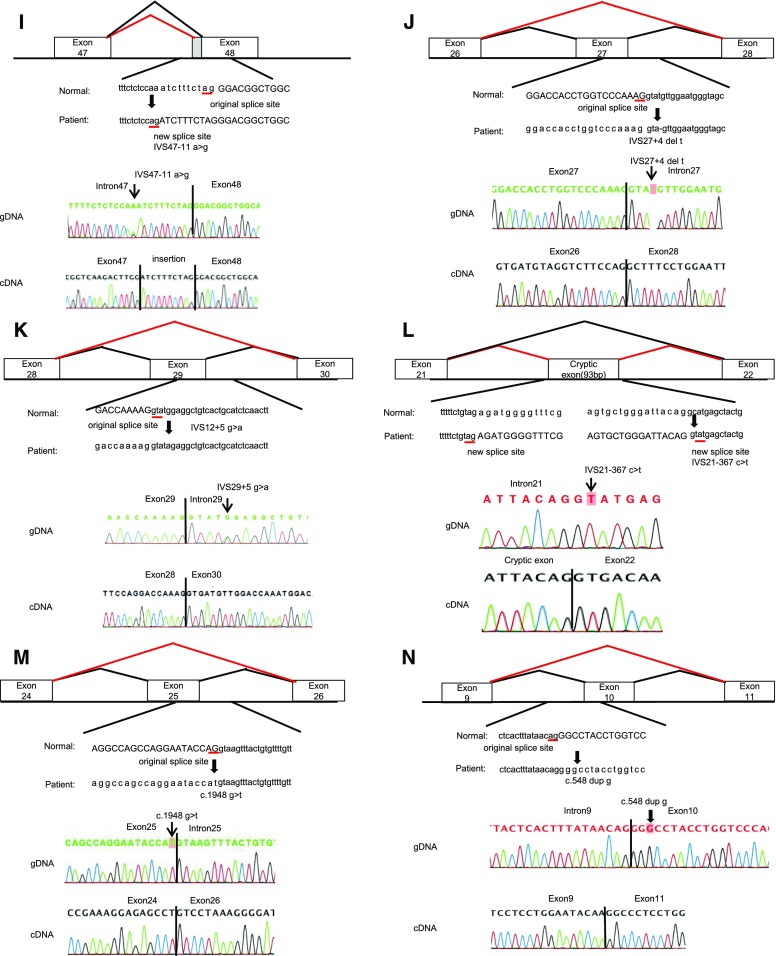
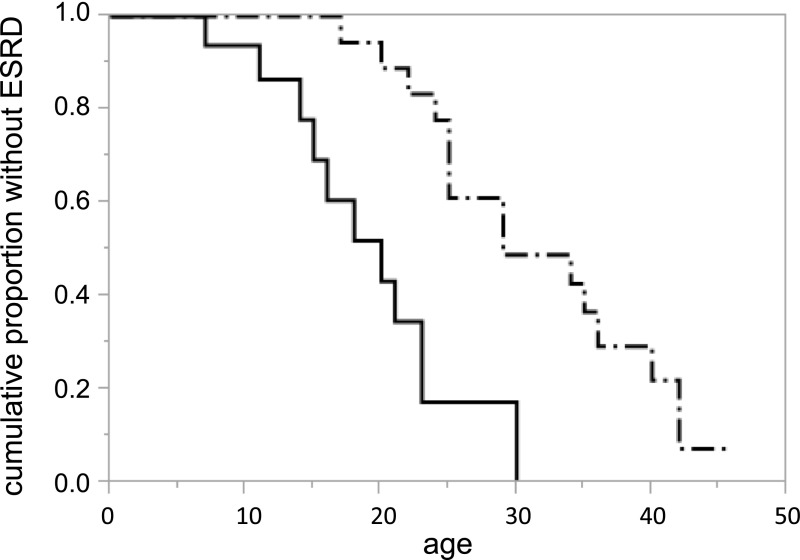
Results: We detected 41 XLAS families with abnormal splicing patterns and described novel XLAS atypical splicing patterns (n=14) other than exon skipping caused by point mutations in the splice consensus sequence. The median age for developing ESRD was 20 years (95% confidence interval, 14 to 23 years) among patients with truncating mutations and 29 years (95% confidence interval, 25 to 40 years) among patients with nontruncating mutations (P=0.001).
Conclusions: We report unpredictable atypical splicing in the COL4A5 gene in male patients with XLAS and reveal that renal prognosis differs significantly for patients with truncating versus nontruncating splicing abnormalities. Our results suggest that splicing modulation should be explored as a therapy for XLAS with truncating mutations.
Keywords: X-linked Alport syndrome; genotype-phenotype correlation; renal prognosis; splicing abnormalities; transcript analysis.
Copyright © 2018 by the American Society of Nephrology.
Figures
References
-
- Kashtan CE: Alport syndrome and thin glomerular basement membrane disease. J Am Soc Nephrol 9: 1736–1750, 1998 - PubMed
-
- Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al. .: X-linked Alport syndrome: Natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol 11: 649–657, 2000 - PubMed
-
- Gross O, Netzer KO, Lambrecht R, Seibold S, Weber M: Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: Impact on clinical counselling. Nephrol Dial Transplant 17: 1218–1227, 2002 - PubMed
-
- Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM: An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 2: 90–95, 1988 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
