

Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement
- PMID: 29959430
- PMCID: PMC6541219
- DOI: 10.1038/s41574-018-0042-0
Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement
Abstract
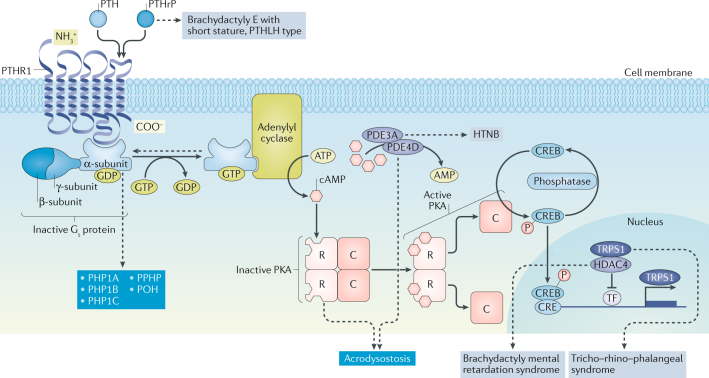
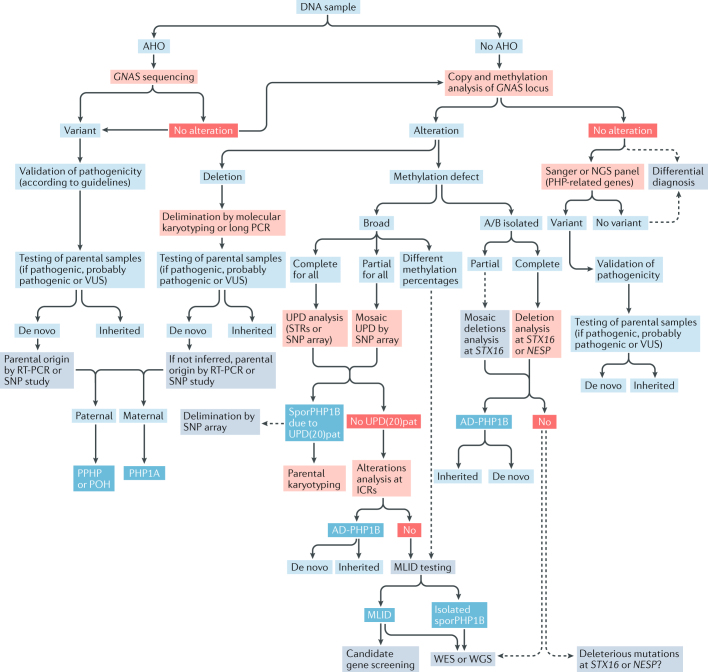
This Consensus Statement covers recommendations for the diagnosis and management of patients with pseudohypoparathyroidism (PHP) and related disorders, which comprise metabolic disorders characterized by physical findings that variably include short bones, short stature, a stocky build, early-onset obesity and ectopic ossifications, as well as endocrine defects that often include resistance to parathyroid hormone (PTH) and TSH. The presentation and severity of PHP and its related disorders vary between affected individuals with considerable clinical and molecular overlap between the different types. A specific diagnosis is often delayed owing to lack of recognition of the syndrome and associated features. The participants in this Consensus Statement agreed that the diagnosis of PHP should be based on major criteria, including resistance to PTH, ectopic ossifications, brachydactyly and early-onset obesity. The clinical and laboratory diagnosis should be confirmed by a molecular genetic analysis. Patients should be screened at diagnosis and during follow-up for specific features, such as PTH resistance, TSH resistance, growth hormone deficiency, hypogonadism, skeletal deformities, oral health, weight gain, glucose intolerance or type 2 diabetes mellitus, and hypertension, as well as subcutaneous and/or deeper ectopic ossifications and neurocognitive impairment. Overall, a coordinated and multidisciplinary approach from infancy through adulthood, including a transition programme, should help us to improve the care of patients affected by these disorders.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Albright F, Burnett CH, Smith PH, Parson W. Pseudohypoparathyroidism — an example of ‘Seabright-Bantam syndrome’. Endocrinology. 1942;30:922–932.
-
- Albright F, Forbes AP, Henneman PH. Pseudo-pseudohypoparathyroidism. Trans. Assoc. Am. Physicians. 1952;65:337–350. - PubMed
-
- Kaplan FS, et al. Progressive osseous heteroplasia: a distinct developmental disorder of heterotopic ossification. Two new case reports and follow-up of three previously reported cases. J. Bone Joint Surg. Am. 1994;76:425–436. - PubMed
-
- Mantovani G, Spada A, Elli FM. Pseudohypoparathyroidism and Gsα-cAMP-linked disorders: current view and open issues. Nat. Rev. Endocrinol. 2016;12:347–356. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
- Archivio Istituzionale della Ricerca Unimi - Access Free Full Text
- Enlighten: Publications, University of Glasgow - Access Free Full Text
- Europe PubMed Central
- Nature Publishing Group
- PubMed Central
- Universidad Autonoma de Madrid Biblos-e Archivo
- University of Turin Instituional Repository AperTO - Free full text
Other Literature Sources
Medical
