

De Novo Truncating Mutations in WASF1 Cause Intellectual Disability with Seizures
- PMID: 29961568
- PMCID: PMC6037130
- DOI: 10.1016/j.ajhg.2018.06.001
De Novo Truncating Mutations in WASF1 Cause Intellectual Disability with Seizures
Abstract
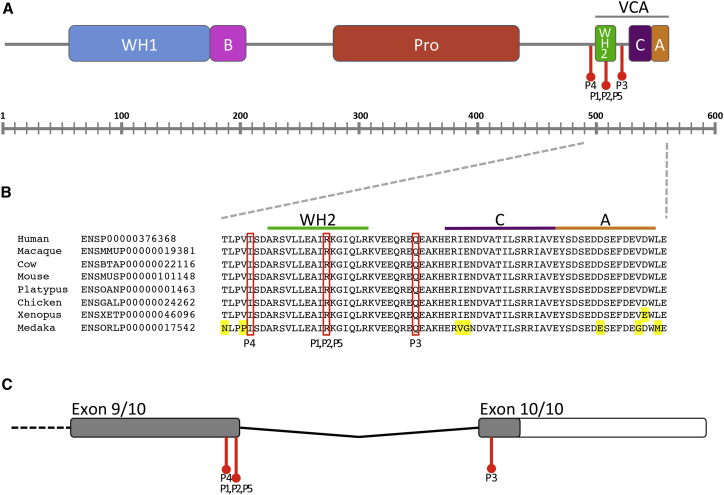
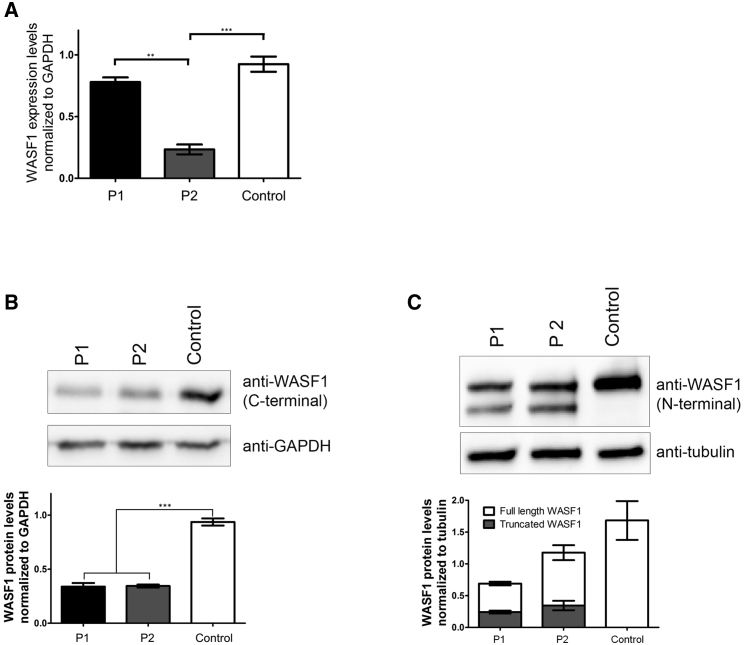
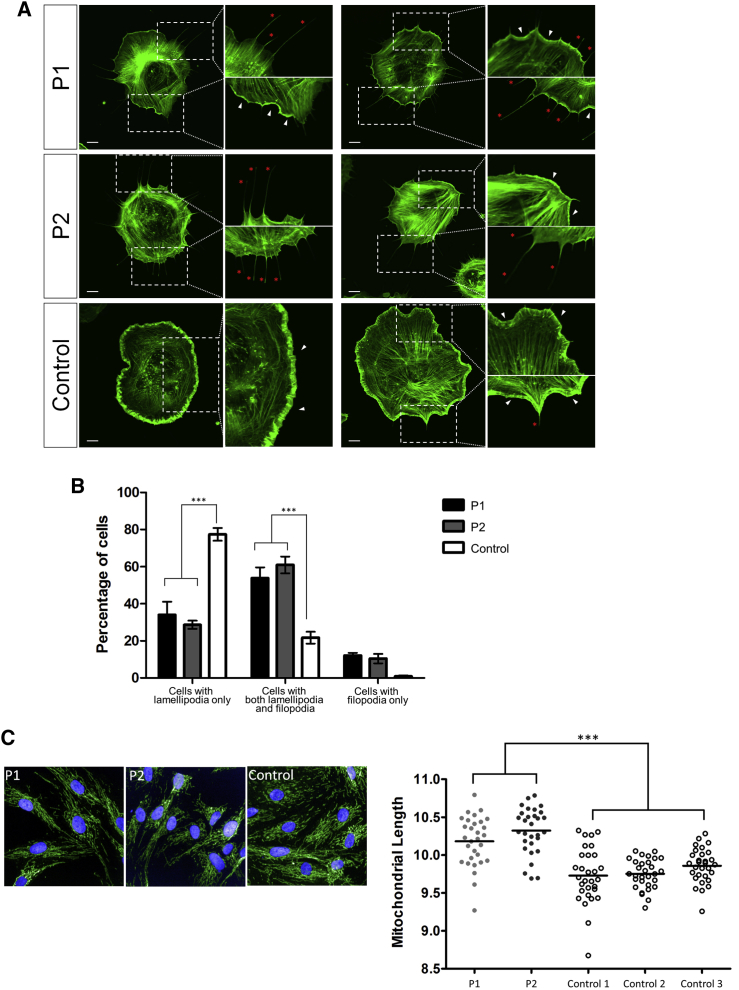
Next-generation sequencing has been invaluable in the elucidation of the genetic etiology of many subtypes of intellectual disability in recent years. Here, using exome sequencing and whole-genome sequencing, we identified three de novo truncating mutations in WAS protein family member 1 (WASF1) in five unrelated individuals with moderate to profound intellectual disability with autistic features and seizures. WASF1, also known as WAVE1, is part of the WAVE complex and acts as a mediator between Rac-GTPase and actin to induce actin polymerization. The three mutations connected by Matchmaker Exchange were c.1516C>T (p.Arg506Ter), which occurs in three unrelated individuals, c.1558C>T (p.Gln520Ter), and c.1482delinsGCCAGG (p.Ile494MetfsTer23). All three variants are predicted to partially or fully disrupt the C-terminal actin-binding WCA domain. Functional studies using fibroblast cells from two affected individuals with the c.1516C>T mutation showed a truncated WASF1 and a defect in actin remodeling. This study provides evidence that de novo heterozygous mutations in WASF1 cause a rare form of intellectual disability.
Keywords: WASF1; WAVE1 complex; actin cytoskeleton; autism; developmental delay; lamellipodia; neurodevelopmental disorder; recurrent de novo truncating mutations; seizures.
Crown Copyright © 2018. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Vissers L.E.L.M., Gilissen C., Veltman J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2016;17:9–18. - PubMed
-
- Martínez F., Caro-Llopis A., Roselló M., Oltra S., Mayo S., Monfort S., Orellana C. High diagnostic yield of syndromic intellectual disability by targeted next-generation sequencing. J. Med. Genet. 2017;54:87–92. - PubMed
-
- Veltman J.A., Brunner H.G. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012;13:565–575. - PubMed
-
- Rauch A., Wieczorek D., Graf E., Wieland T., Endele S., Schwarzmayr T., Albrecht B., Bartholdi D., Beygo J., Di Donato N. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
