

Recovering Gene Interactions from Single-Cell Data Using Data Diffusion
- PMID: 29961576
- PMCID: PMC6771278
- DOI: 10.1016/j.cell.2018.05.061
Recovering Gene Interactions from Single-Cell Data Using Data Diffusion
Abstract
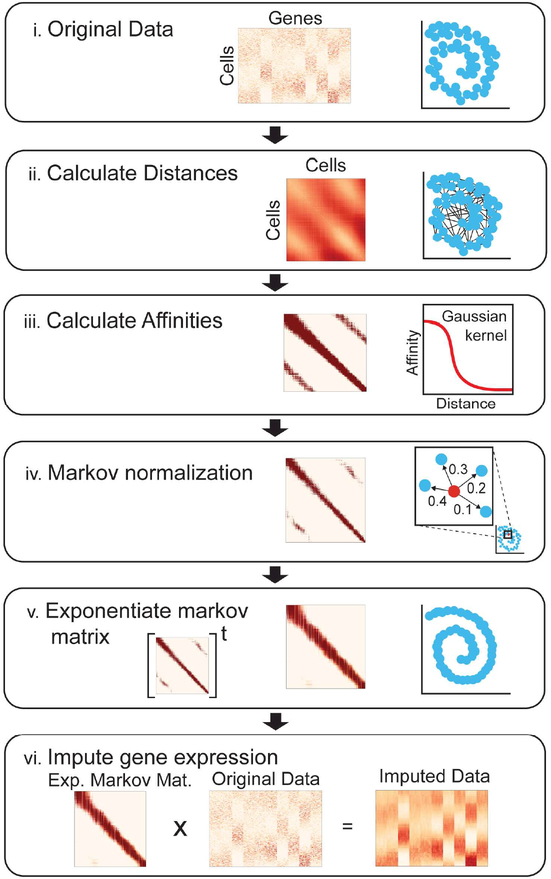
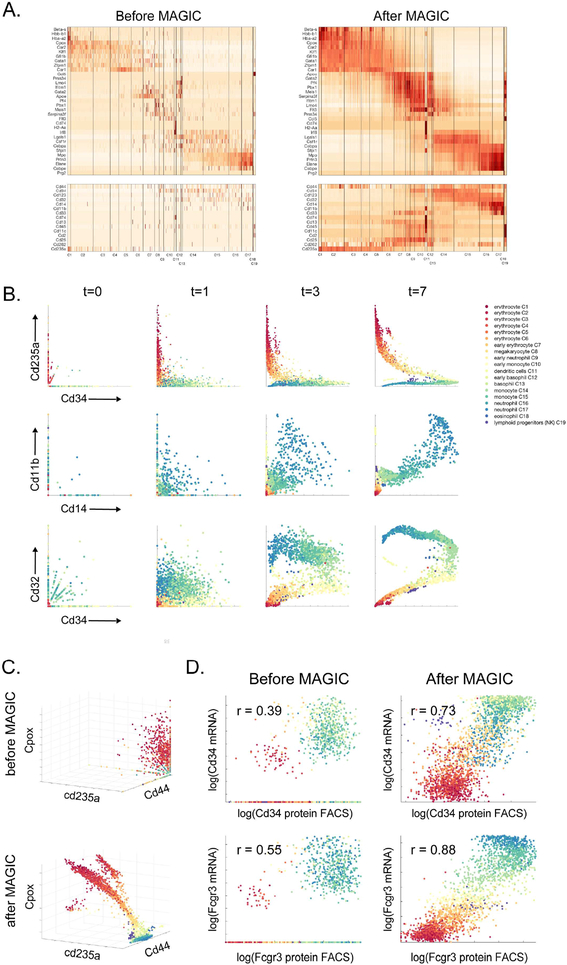
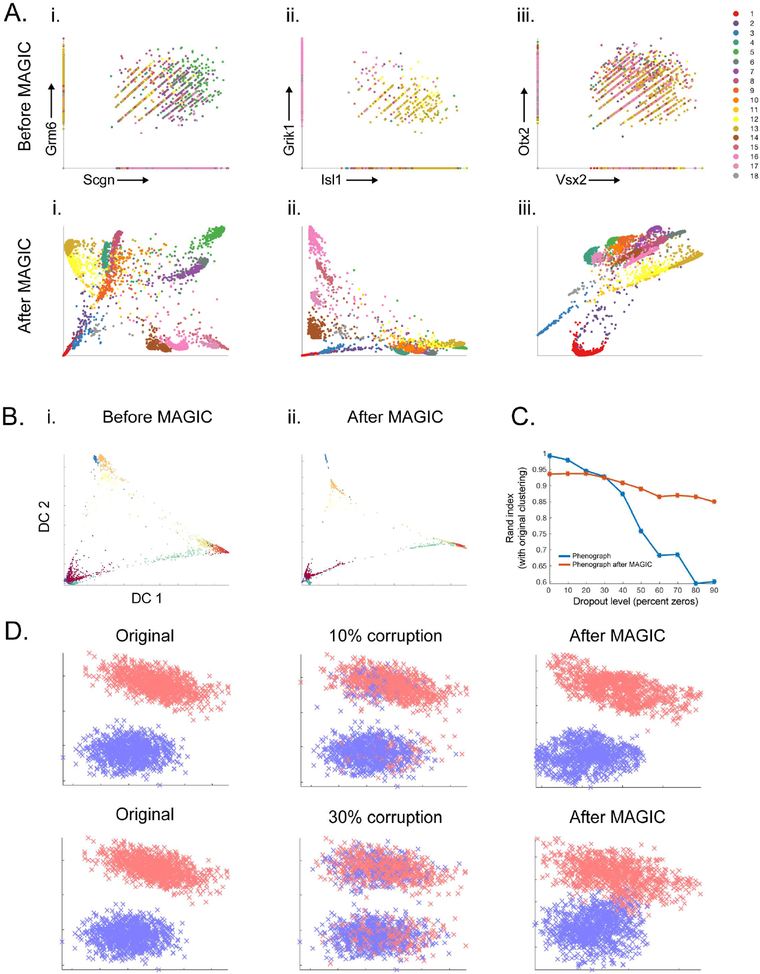
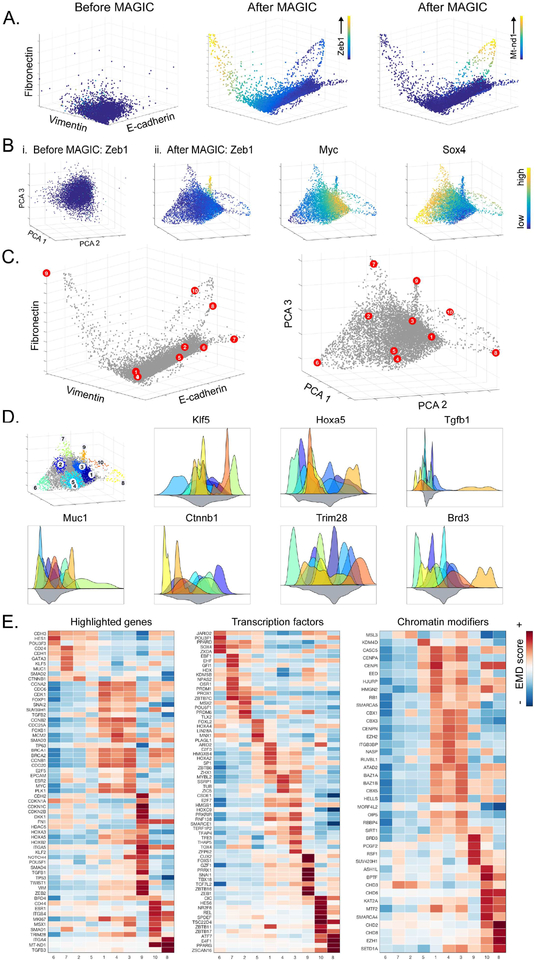
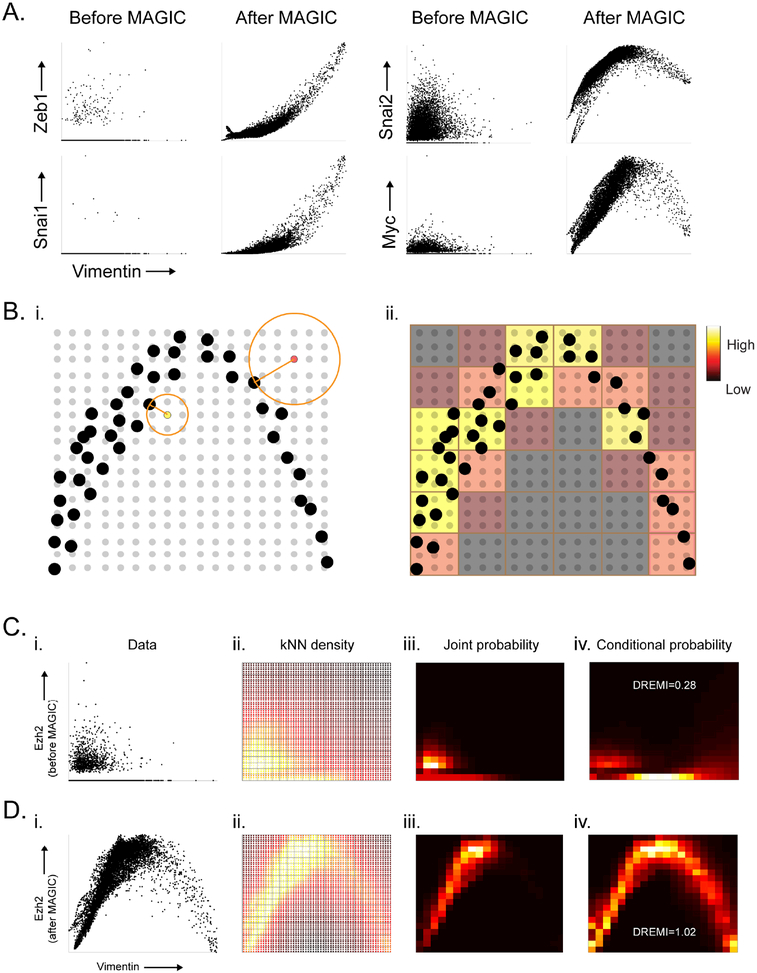
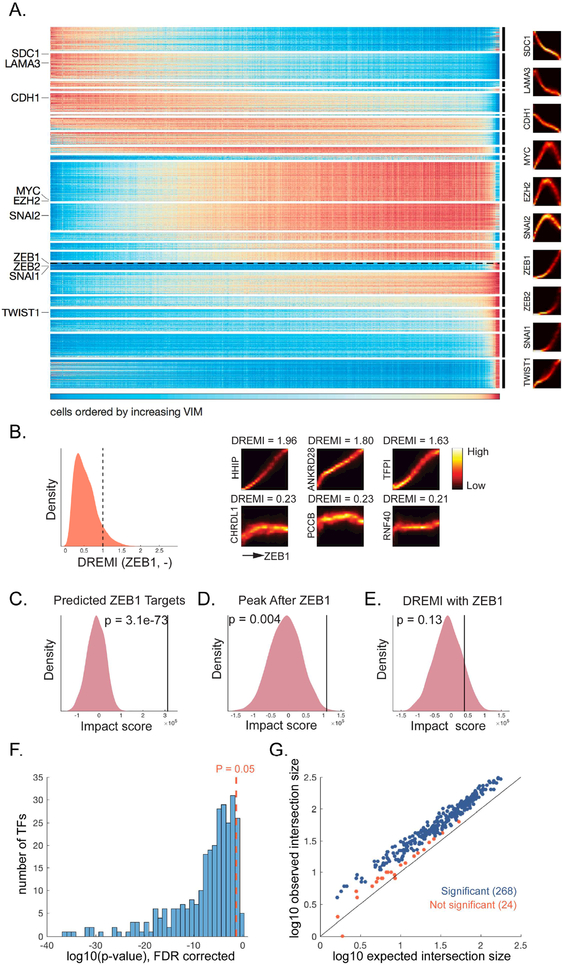
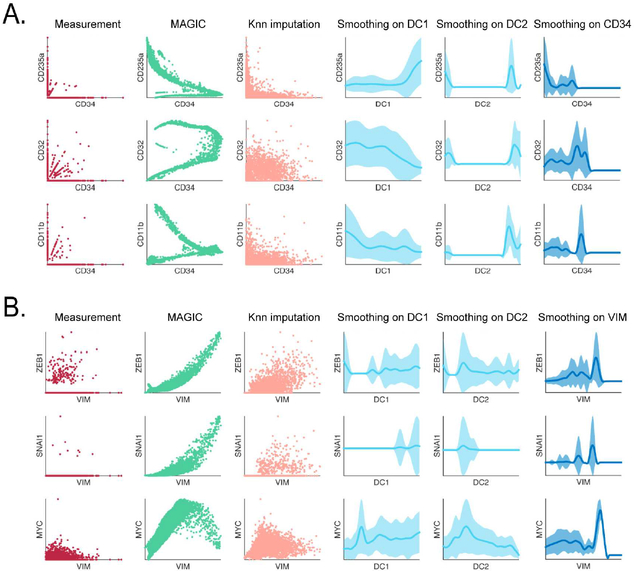
Single-cell RNA sequencing technologies suffer from many sources of technical noise, including under-sampling of mRNA molecules, often termed "dropout," which can severely obscure important gene-gene relationships. To address this, we developed MAGIC (Markov affinity-based graph imputation of cells), a method that shares information across similar cells, via data diffusion, to denoise the cell count matrix and fill in missing transcripts. We validate MAGIC on several biological systems and find it effective at recovering gene-gene relationships and additional structures. Applied to the epithilial to mesenchymal transition, MAGIC reveals a phenotypic continuum, with the majority of cells residing in intermediate states that display stem-like signatures, and infers known and previously uncharacterized regulatory interactions, demonstrating that our approach can successfully uncover regulatory relations without perturbations.
Keywords: EMT; imputation; manifold learning; regulatory networks; single-cell RNA sequencing.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures
References
-
- Achlioptas D, and McSherry F (2007). Fast computation of low-rank matrix approximations. Journal of the ACM (JACM) 54, 9.
-
- Botev ZI, Grotowski JF, and Kroese DP (2010). Kernel Density Estimation Via Diffusion. Annals of Statistics 38, 2916–2957.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
