

Oxidized phospholipid signaling in traumatic brain injury
- PMID: 29964171
- PMCID: PMC6098726
- DOI: 10.1016/j.freeradbiomed.2018.06.031
Oxidized phospholipid signaling in traumatic brain injury
Abstract
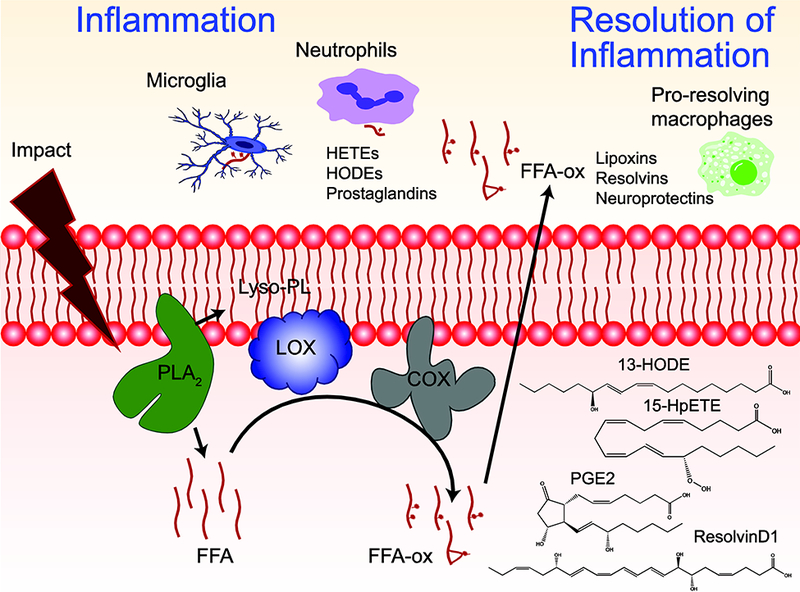
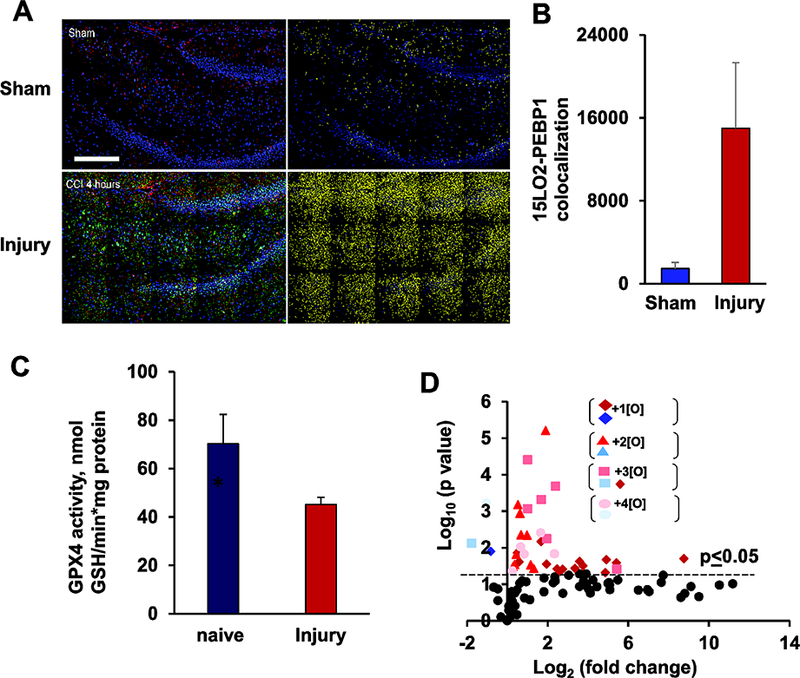
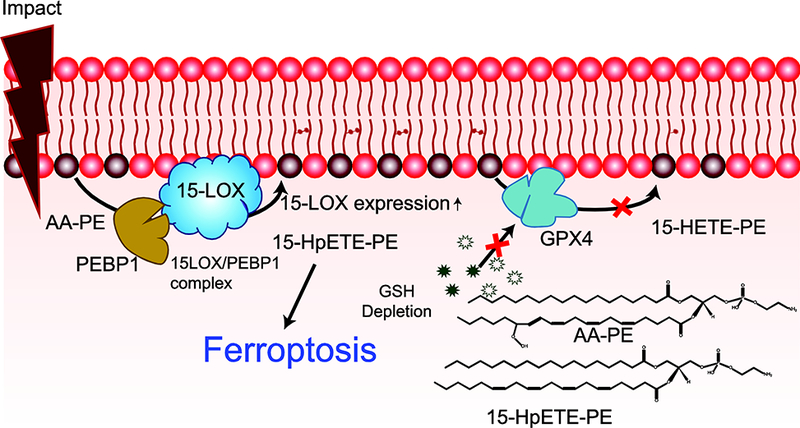
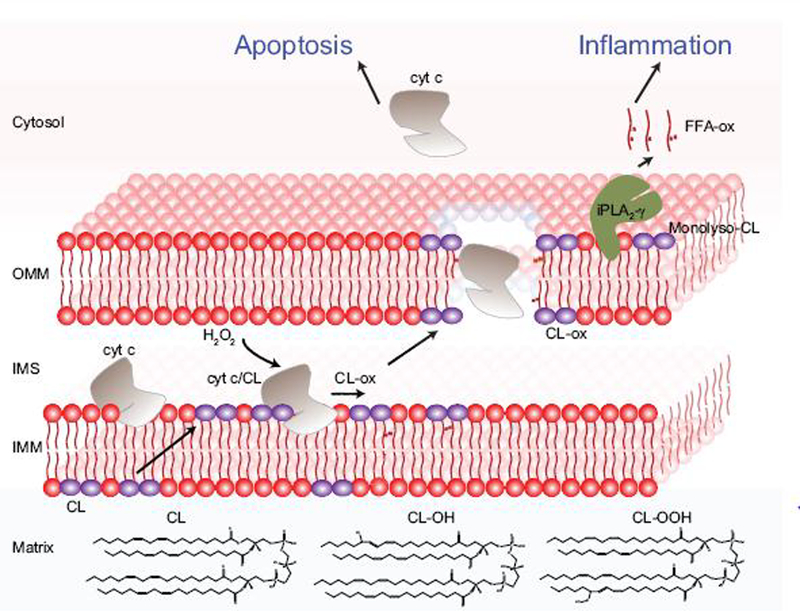
Oxidative stress is a major contributor to secondary injury signaling cascades following traumatic brain injury (TBI). The role of lipid peroxidation in the pathophysiology of a traumatic insult to neural tissue is increasingly recognized. As the methods to quantify lipid peroxidation have gradually improved, so has the understanding of mechanistic details of lipid peroxidation and related signaling events in the injury pathogenesis. While free-radical mediated, non-enzymatic lipid peroxidation has long been studied, recent advances in redox lipidomics have demonstrated the significant contribution of enzymatic lipid peroxidation to TBI pathogenesis. Complex interactions between inflammation, phospholipid peroxidation, and hydrolysis define the engagement of different cell death programs and the severity of injury and outcome. This review focuses on enzymatic phospholipid peroxidation after TBI, including the mechanism of production, signaling roles in secondary injury pathology, and temporal course of production with respect to inflammatory response. In light of the newly identified phospholipid oxidation mechanisms, we also discuss possible therapeutic targets to improve neurocognitive outcome after TBI. Finally, we discuss current limitations in identifying oxidized phospholipids and possible methodologic improvements that can offer a deeper insight into the region-specific distribution and subcellular localization of phospholipid oxidation after TBI.
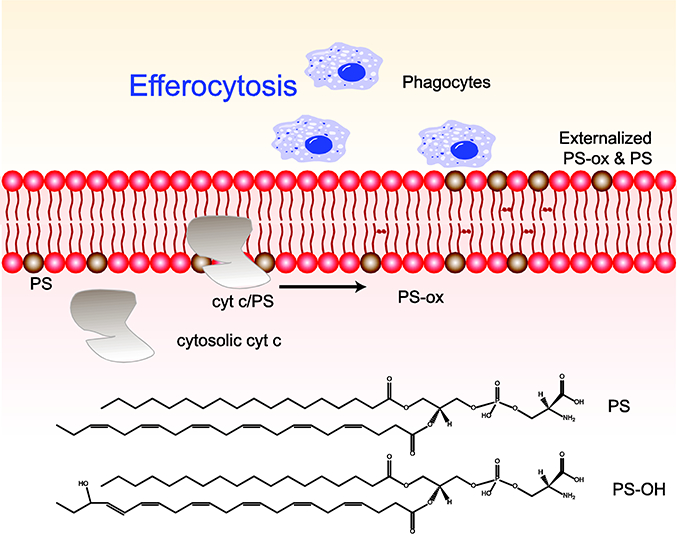
Keywords: Apoptosis; Efferocytosis; Ferroptosis; Inflammation; Lipid mediator; Redox lipidomics.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Mitochondrial damage & lipid signaling in traumatic brain injury.Exp Neurol. 2020 Jul;329:113307. doi: 10.1016/j.expneurol.2020.113307. Epub 2020 Apr 11. Exp Neurol. 2020. PMID: 32289317 Free PMC article. Review.
-
Redox Epiphospholipidome in Programmed Cell Death Signaling: Catalytic Mechanisms and Regulation.Front Endocrinol (Lausanne). 2021 Feb 19;11:628079. doi: 10.3389/fendo.2020.628079. eCollection 2020. Front Endocrinol (Lausanne). 2021. PMID: 33679610 Free PMC article. Review.
-
Global assessment of oxidized free fatty acids in brain reveals an enzymatic predominance to oxidative signaling after trauma.Biochim Biophys Acta Mol Basis Dis. 2017 Oct;1863(10 Pt B):2601-2613. doi: 10.1016/j.bbadis.2017.03.015. Epub 2017 Mar 25. Biochim Biophys Acta Mol Basis Dis. 2017. PMID: 28347845 Free PMC article.
-
Therapies targeting lipid peroxidation in traumatic brain injury.Brain Res. 2016 Jun 1;1640(Pt A):57-76. doi: 10.1016/j.brainres.2016.02.006. Epub 2016 Feb 10. Brain Res. 2016. PMID: 26872597 Free PMC article. Review.
-
Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis.Ann Neurol. 2007 Aug;62(2):154-69. doi: 10.1002/ana.21168. Ann Neurol. 2007. PMID: 17685468
Cited by
-
Prokineticin-2 prevents neuronal cell deaths in a model of traumatic brain injury.Nat Commun. 2021 Jul 9;12(1):4220. doi: 10.1038/s41467-021-24469-y. Nat Commun. 2021. PMID: 34244497 Free PMC article.
-
Truncated oxidized phospholipids exacerbate endothelial dysfunction and lung injury caused by bacterial pathogens.Cell Signal. 2023 Sep;109:110804. doi: 10.1016/j.cellsig.2023.110804. Epub 2023 Jul 10. Cell Signal. 2023. PMID: 37437826 Free PMC article.
-
Edaravone Alleviates Traumatic Brain Injury by Inhibition of Ferroptosis via FSP1 Pathway.Mol Neurobiol. 2024 Dec;61(12):10448-10461. doi: 10.1007/s12035-024-04216-2. Epub 2024 May 11. Mol Neurobiol. 2024. PMID: 38733490 Free PMC article.
-
Mechanisms of Ferritinophagy and Ferroptosis in Diseases.Mol Neurobiol. 2024 Mar;61(3):1605-1626. doi: 10.1007/s12035-023-03640-0. Epub 2023 Sep 22. Mol Neurobiol. 2024. PMID: 37736794 Review.
-
Activation of the Hedgehog Pathway Promotes Recovery of Neurological Function After Traumatic Brain Injury by Protecting the Neurovascular Unit.Transl Stroke Res. 2020 Aug;11(4):720-733. doi: 10.1007/s12975-019-00771-2. Epub 2020 Jan 2. Transl Stroke Res. 2020. PMID: 31898187
References
-
- Faul M, Xu L, Wald MM, Coronado V, Dellinger AM, Traumatic brain injury in the United States: national estimates of prevalence and incidence, 2002–2006, Injury Prevention 16(Suppl 1) (2011) A268.
-
- Coronado VG, Xu L, Basavaraju SV, McGuire LC, Wald MM, Faul MD, Guzman BR, Hemphill JD, Surveillance for traumatic brain injury-related deaths-United States, 1997–2007, Morbidity and mortality weekly report. Surveillance summaries (Washington, D.C. : 2002) 60(5) (2011) 1–32. - PubMed
-
- Maas AIR, Stocchetti N, Bullock R, Moderate and severe traumatic brain injury in adults, The Lancet Neurology 7(8) (2008) 728–741. - PubMed
-
- Andelic N, Hammergren N, Bautz-Holter E, Sveen U, Brunborg C, Roe C, Functional outcome and health-related quality of life 10 years after moderate-to-severe traumatic brain injury, Acta neurologica Scandinavica 120(1) (2009) 16–23. - PubMed
-
- Andriessen TM, Horn J, Franschman G, van der Naalt J, Haitsma I, Jacobs B, Steyerberg EW, Vos PE, Epidemiology, severity classification, and outcome of moderate and severe traumatic brain injury: a prospective multicenter study, Journal of neurotrauma 28(10) (2011) 2019–31. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
