

TGF-β signaling alters H4K20me3 status via miR-29 and contributes to cellular senescence and cardiac aging
- PMID: 29967491
- PMCID: PMC6028646
- DOI: 10.1038/s41467-018-04994-z
TGF-β signaling alters H4K20me3 status via miR-29 and contributes to cellular senescence and cardiac aging
Abstract
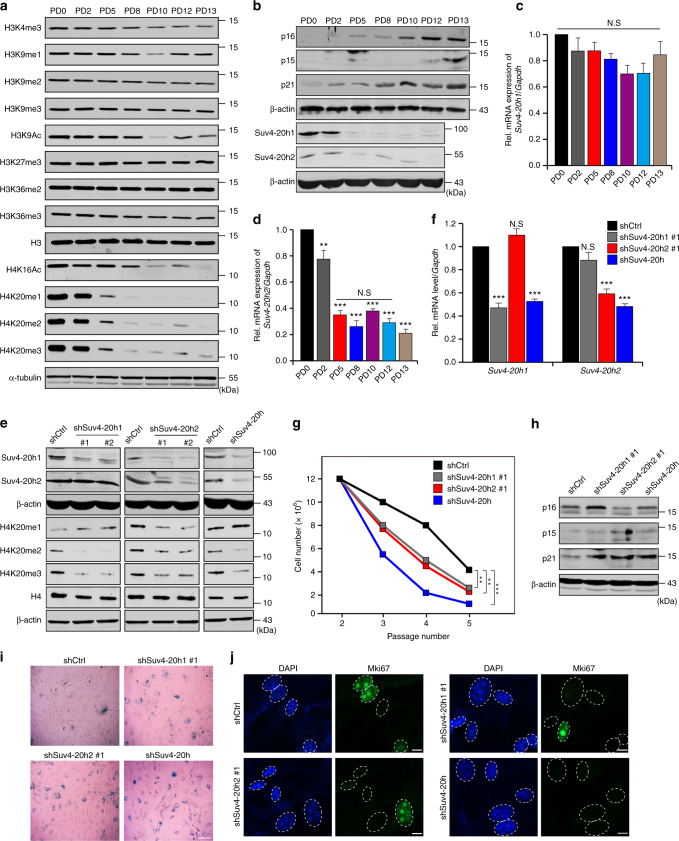
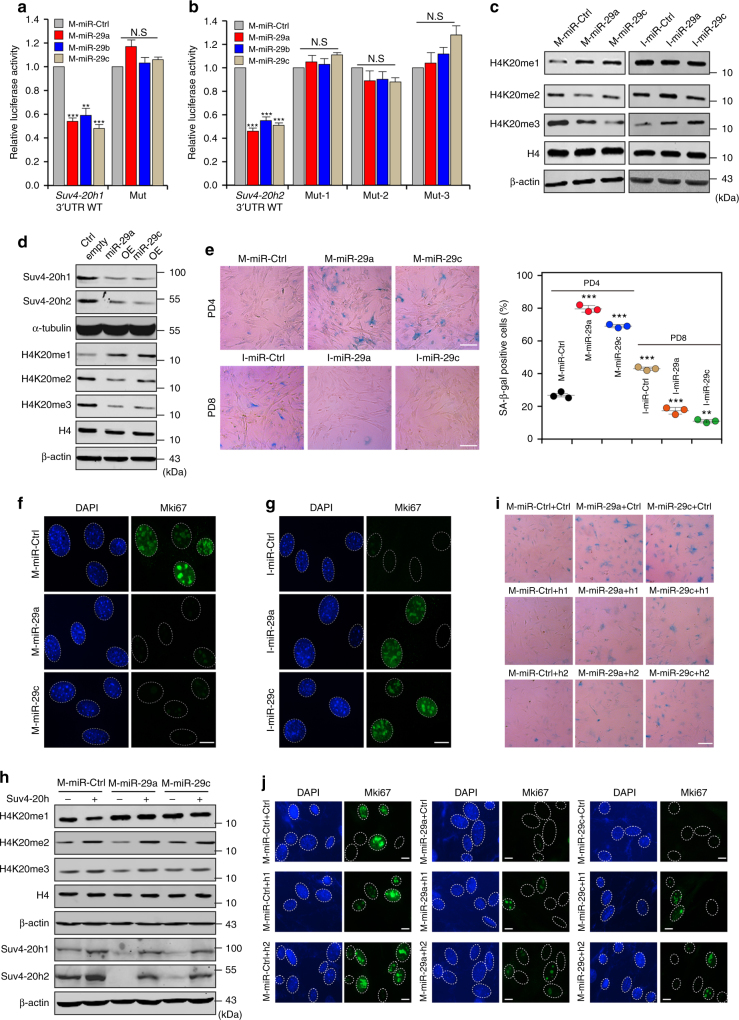
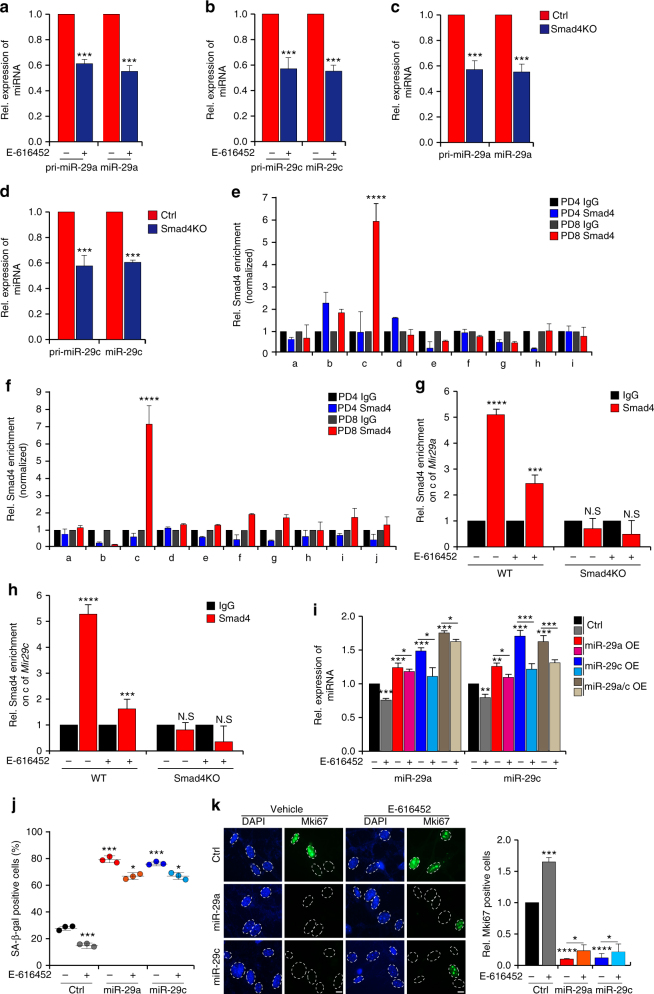
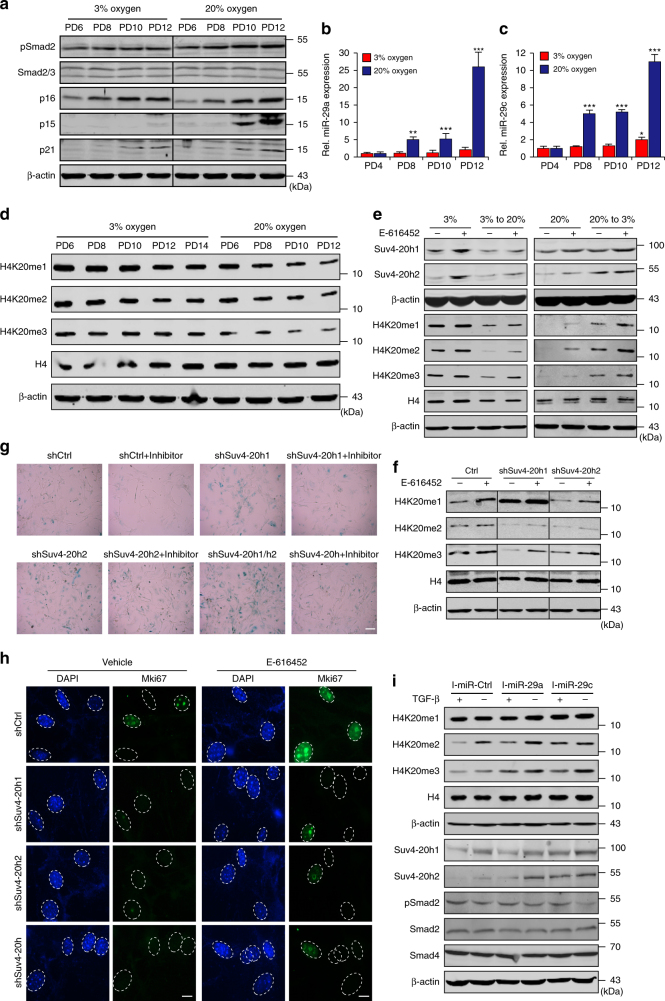
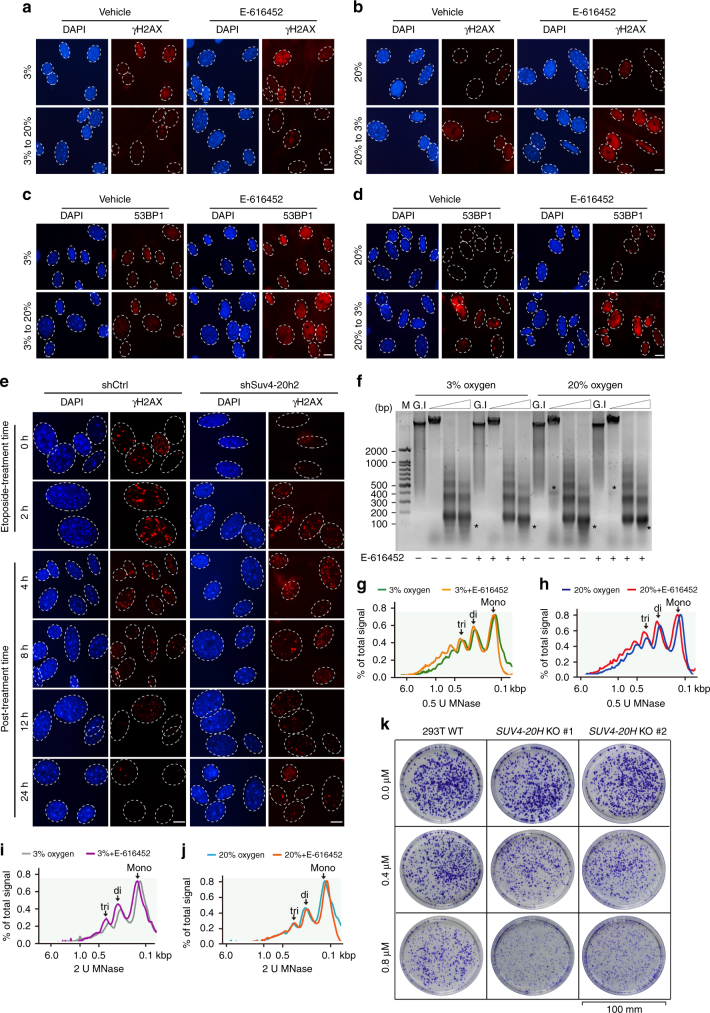
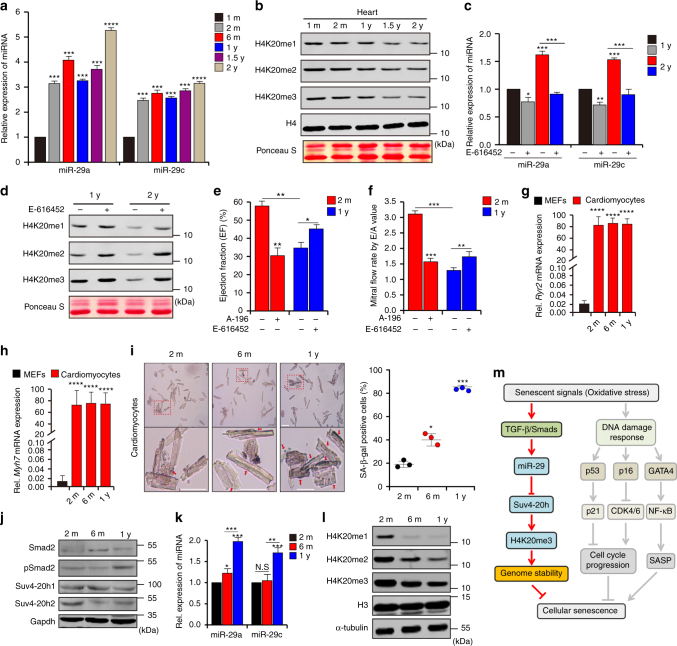
Cellular senescence is a well-orchestrated programmed process involved in age-related pathologies, tumor suppression and embryonic development. TGF-β/Smad is one of the predominant pathways that regulate damage-induced and developmentally programmed senescence. Here we show that canonical TGF-β signaling promotes senescence via miR-29-induced loss of H4K20me3. Mechanistically, oxidative stress triggers TGF-β signaling. Activated TGF-β signaling gives rise to acute accumulation of miR-29a and miR-29c, both of which directly suppress their novel target, Suv4-20h, thus reducing H4K20me3 abundance in a Smad-dependent manner, which compromises DNA damage repair and genome maintenance. Loss of H4K20me3 mediated by the senescent TGF-β/miR-29 pathway contributes to cardiac aging in vivo. Disruption of TGF-β signaling restores H4K20me3 and improves cardiac function in aged mice. Our study highlights the sequential mechanisms underlying the regulation of senescence, from senescence-inducing triggers to activation of responsive signaling followed by specific epigenetic alterations, shedding light on potential therapeutic interventions in cardiac aging.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- 31471205/National Natural Science Foundation of China (National Science Foundation of China)/International
- 31171255/National Natural Science Foundation of China (National Science Foundation of China)/International
- 91219101/National Natural Science Foundation of China (National Science Foundation of China)/International
- 81330009/National Natural Science Foundation of China (National Science Foundation of China)/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
