

CD4+ T Cell Fate Decisions Are Stochastic, Precede Cell Division, Depend on GITR Co-Stimulation, and Are Associated With Uropodium Development
- PMID: 29967616
- PMCID: PMC6015874
- DOI: 10.3389/fimmu.2018.01381
CD4+ T Cell Fate Decisions Are Stochastic, Precede Cell Division, Depend on GITR Co-Stimulation, and Are Associated With Uropodium Development
Abstract
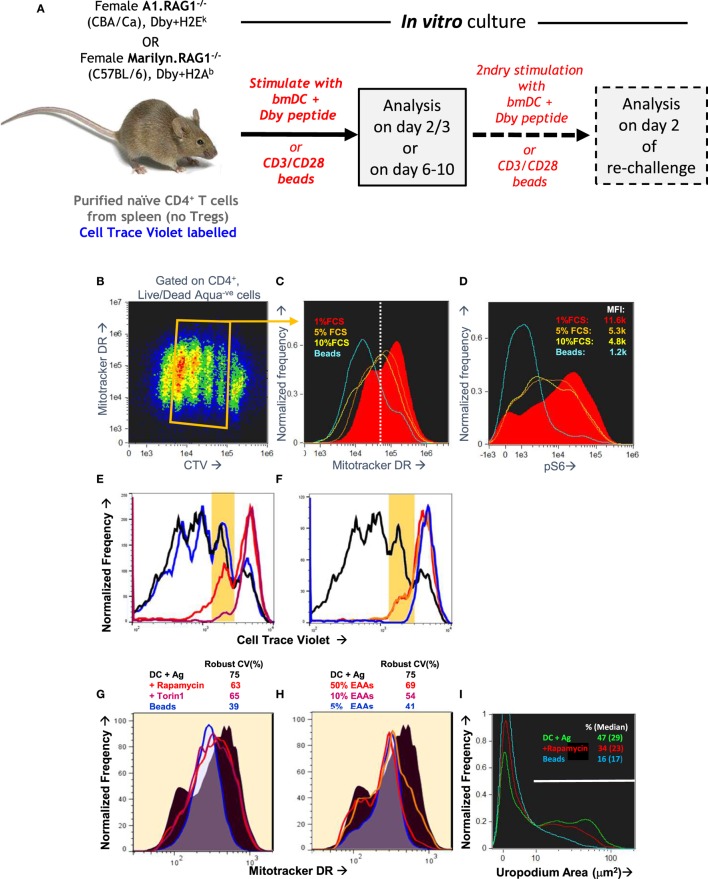
During an immune response, naïve CD4+ T cells proliferate and generate a range of effector, memory, and regulatory T cell subsets, but how these processes are co-ordinated remains unclear. A traditional model suggests that memory cells use mitochondrial respiration and are survivors from a pool of previously proliferating and glycolytic, but short-lived effector cells. A more recent model proposes a binary commitment to either a memory or effector cell lineage during a first, asymmetric cell division, with each lineage able to undergo subsequent proliferation and differentiation. We used improved fixation and staining methods with imaging flow cytometry in an optimized in vitro system that indicates a third model. We found that cell fates result from stochastic decisions that depend on GITR co-stimulation and which take place before any cell division. Effector cell commitment is associated with mTORC2 signaling leading to uropodium development, while developing memory cells lose mitochondria, have a nuclear localization of NFκB and depend on TGFβ for their survival. Induced, T helper subsets and foxp3+ regulatory T cells were found in both the effector and memory cell lineages. This in vitro model of T cell differentiation is well suited to testing how manipulation of cytokine, nutrient, and other components of the microenvironment might be exploited for therapeutic purposes.
Keywords: GITR; T cell differentiation; asymmetric cell division; cell fate; imaging flow cytometry; mTOR signaling; uropodium.
Figures
















Similar articles
-
Regulation of Asymmetric Division by Atypical Protein Kinase C Influences Early Specification of CD8(+) T Lymphocyte Fates.Sci Rep. 2016 Jan 14;6:19182. doi: 10.1038/srep19182. Sci Rep. 2016. PMID: 26765121 Free PMC article.
-
The Fate Choice Between Effector and Memory T Cell Lineages: Asymmetry, Signal Integration, and Feedback to Create Bistability.Adv Immunol. 2018;137:43-82. doi: 10.1016/bs.ai.2017.12.003. Epub 2018 Feb 1. Adv Immunol. 2018. PMID: 29455847 Review.
-
GITR differentially affects lung effector T cell subpopulations during influenza virus infection.J Leukoc Biol. 2020 Jun;107(6):953-970. doi: 10.1002/JLB.4AB1219-254R. Epub 2020 Mar 3. J Leukoc Biol. 2020. PMID: 32125017
-
Asymmetric Cell Division in T Lymphocyte Fate Diversification.Trends Immunol. 2015 Nov;36(11):670-683. doi: 10.1016/j.it.2015.09.004. Epub 2015 Oct 20. Trends Immunol. 2015. PMID: 26474675 Free PMC article. Review.
-
GITR pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability.Cancer Immunol Res. 2013 Nov;1(5):320-31. doi: 10.1158/2326-6066.CIR-13-0086. Cancer Immunol Res. 2013. PMID: 24416730 Free PMC article.
Cited by
-
A selective LIS1 requirement for mitotic spindle assembly discriminates distinct T-cell division mechanisms within the T-cell lineage.Elife. 2022 Dec 15;11:e80277. doi: 10.7554/eLife.80277. Elife. 2022. PMID: 36519536 Free PMC article.
-
Model-Based Assessment of the Role of Uneven Partitioning of Molecular Content on Heterogeneity and Regulation of Differentiation in CD8 T-Cell Immune Responses.Front Immunol. 2019 Feb 19;10:230. doi: 10.3389/fimmu.2019.00230. eCollection 2019. Front Immunol. 2019. PMID: 30842771 Free PMC article.
-
Stochastic asymmetric repartition of lytic machinery in dividing CD8+ T cells generates heterogeneous killing behavior.Elife. 2021 Jan 11;10:e62691. doi: 10.7554/eLife.62691. Elife. 2021. PMID: 33427199 Free PMC article.
-
Asymmetric T-cell division: insights from cutting-edge experimental techniques and implications for immunotherapy.Front Immunol. 2024 Mar 1;15:1301378. doi: 10.3389/fimmu.2024.1301378. eCollection 2024. Front Immunol. 2024. PMID: 38495874 Free PMC article. Review.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous