

A senataxin-associated exonuclease SAN1 is required for resistance to DNA interstrand cross-links
- PMID: 29968717
- PMCID: PMC6030175
- DOI: 10.1038/s41467-018-05008-8
A senataxin-associated exonuclease SAN1 is required for resistance to DNA interstrand cross-links
Abstract
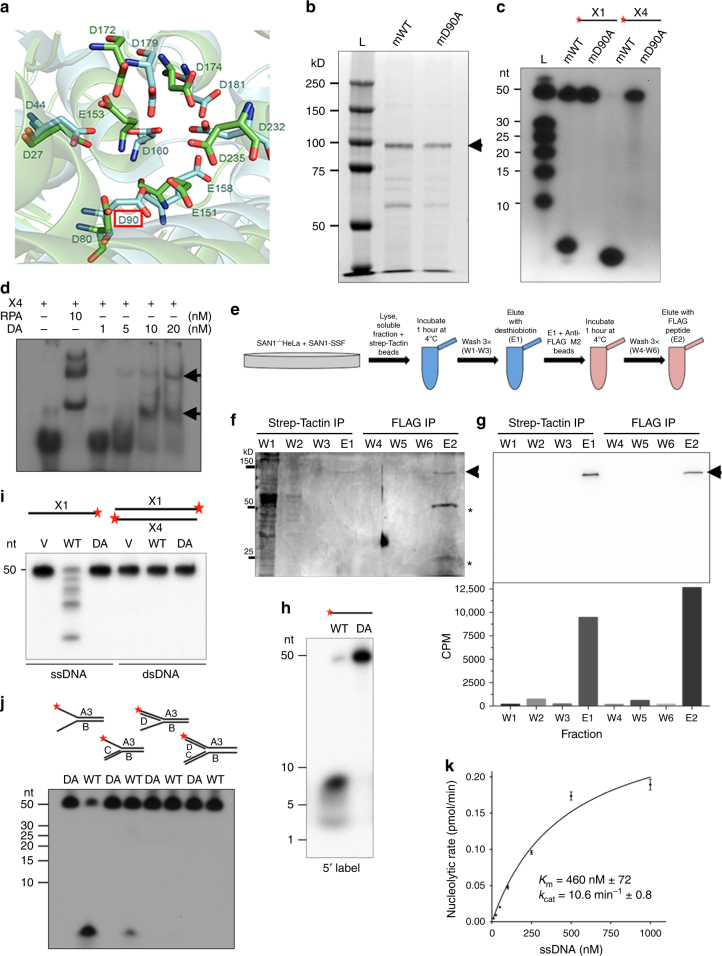
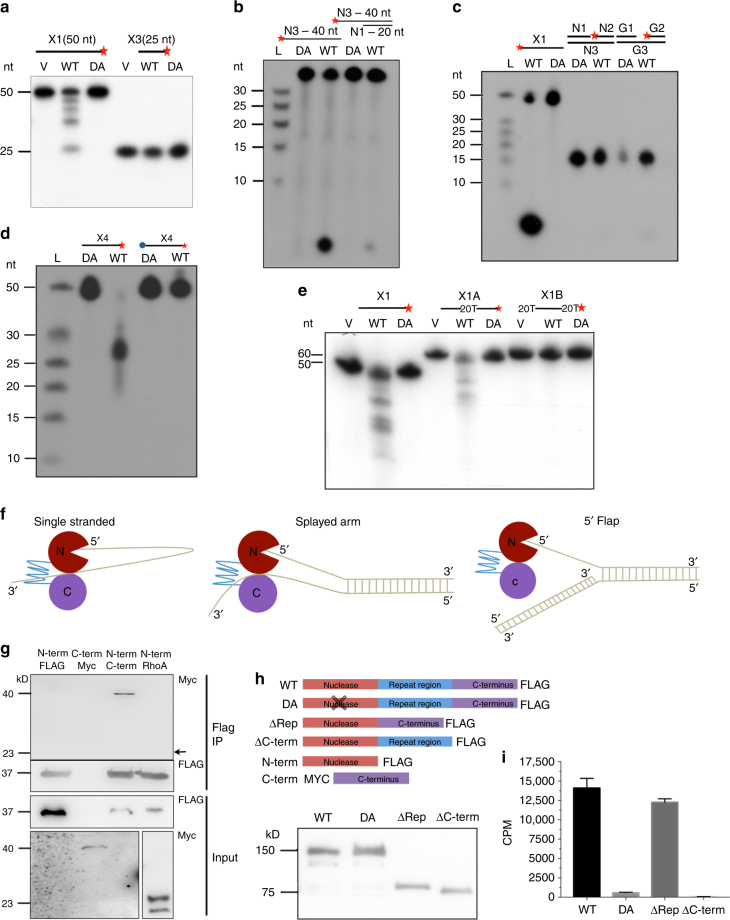
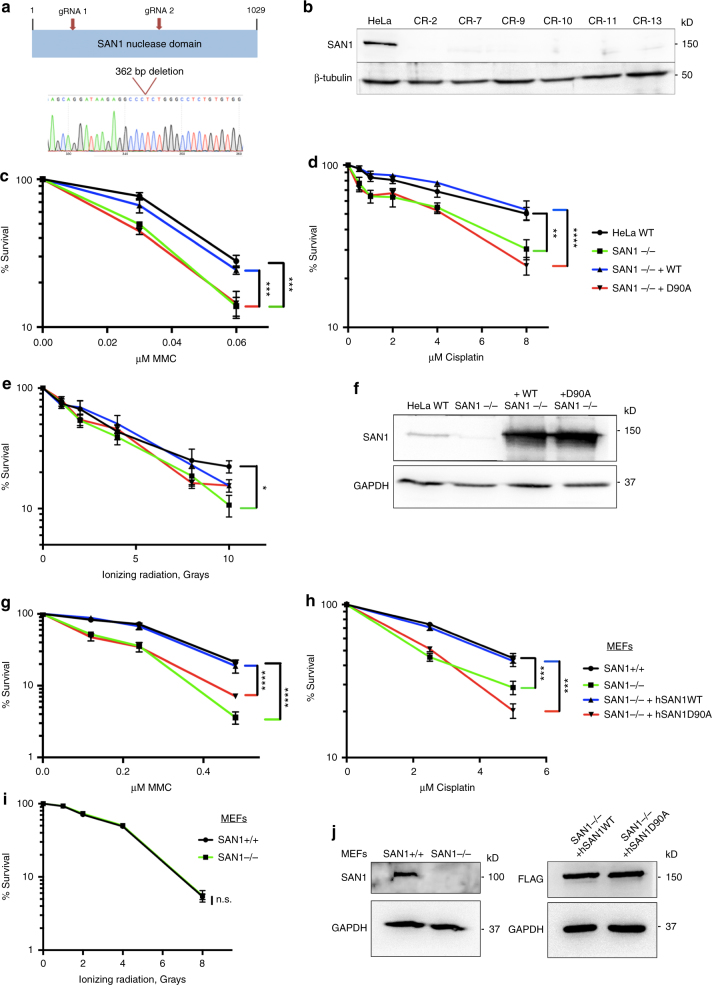
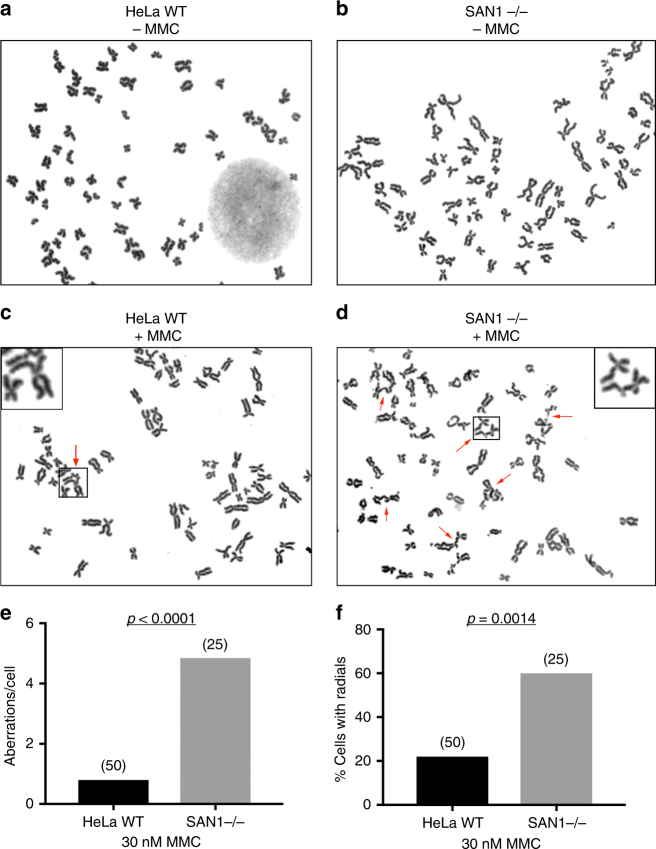
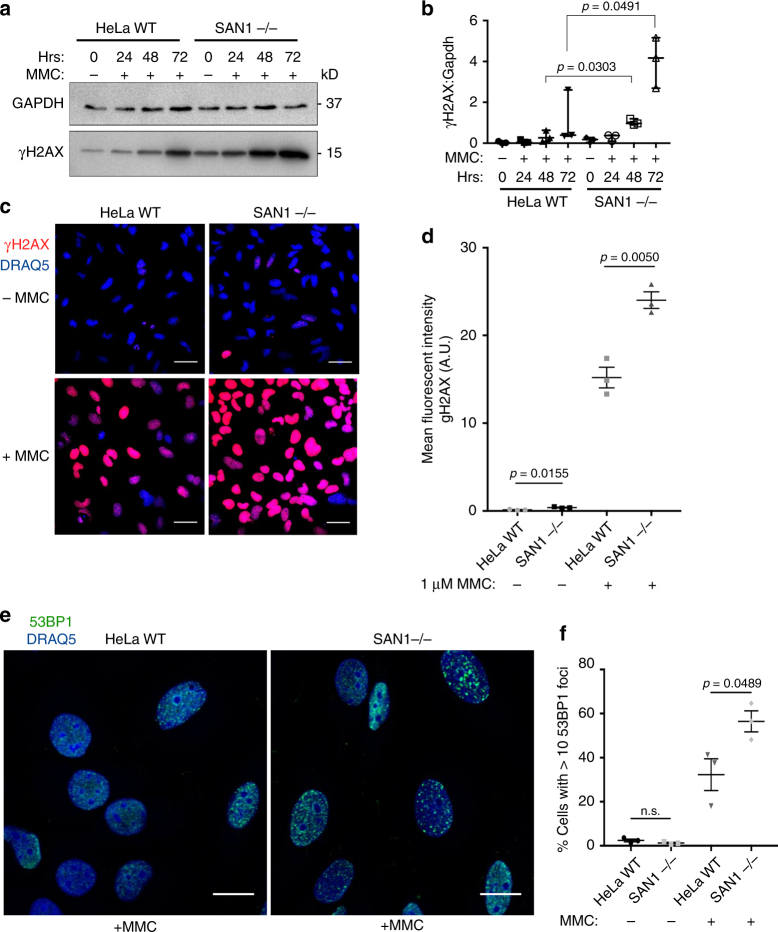
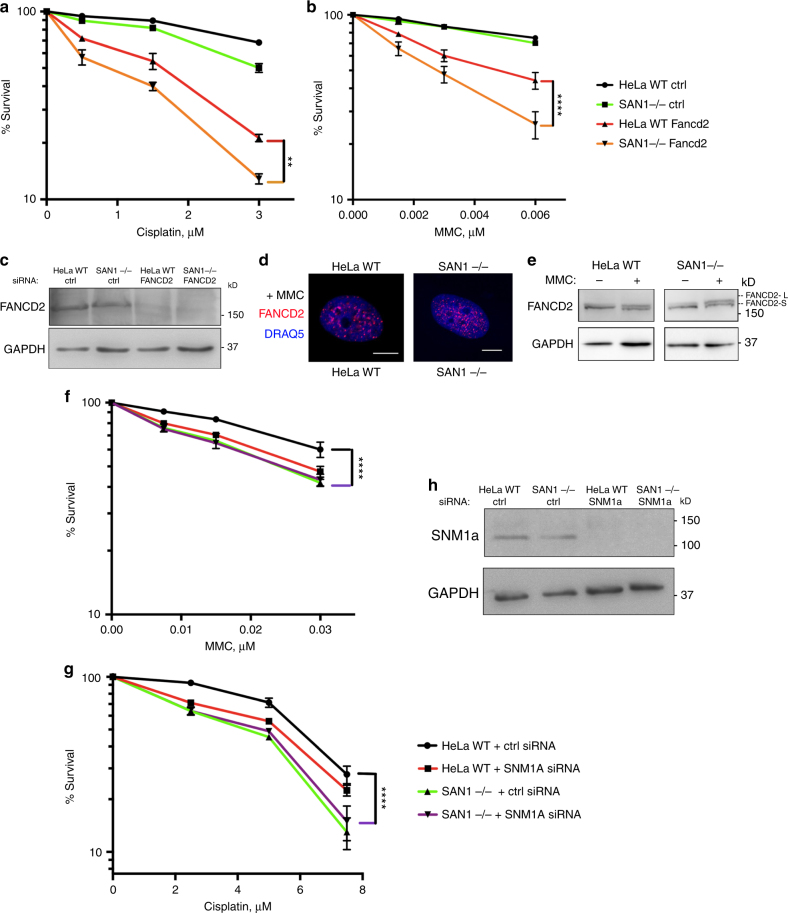
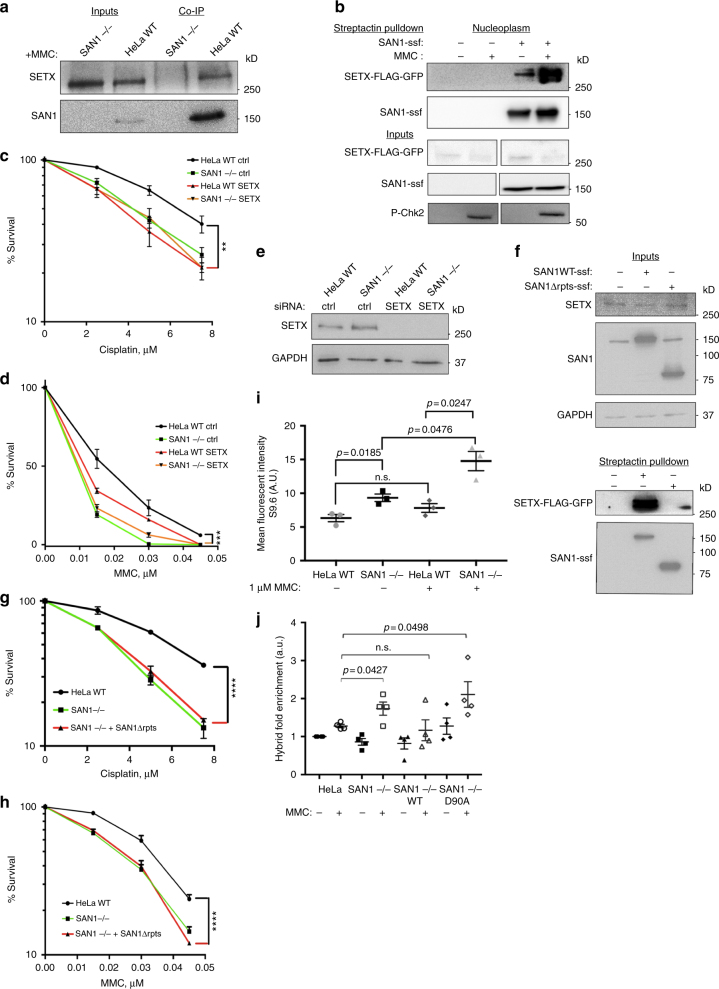
Interstrand DNA cross-links (ICLs) block both replication and transcription, and are commonly repaired by the Fanconi anemia (FA) pathway. However, FA-independent repair mechanisms of ICLs remain poorly understood. Here we report a previously uncharacterized protein, SAN1, as a 5' exonuclease that acts independently of the FA pathway in response to ICLs. Deletion of SAN1 in HeLa cells and mouse embryonic fibroblasts causes sensitivity to ICLs, which is prevented by re-expression of wild type but not nuclease-dead SAN1. SAN1 deletion causes DNA damage and radial chromosome formation following treatment with Mitomycin C, phenocopying defects in the FA pathway. However, SAN1 deletion is not epistatic with FANCD2, a core FA pathway component. Unexpectedly, SAN1 binds to Senataxin (SETX), an RNA/DNA helicase that resolves R-loops. SAN1-SETX binding is increased by ICLs, and is required to prevent cross-link sensitivity. We propose that SAN1 functions with SETX in a pathway necessary for resistance to ICLs.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
