

A selective high affinity MYC-binding compound inhibits MYC:MAX interaction and MYC-dependent tumor cell proliferation
- PMID: 29968736
- PMCID: PMC6030159
- DOI: 10.1038/s41598-018-28107-4
A selective high affinity MYC-binding compound inhibits MYC:MAX interaction and MYC-dependent tumor cell proliferation
Abstract
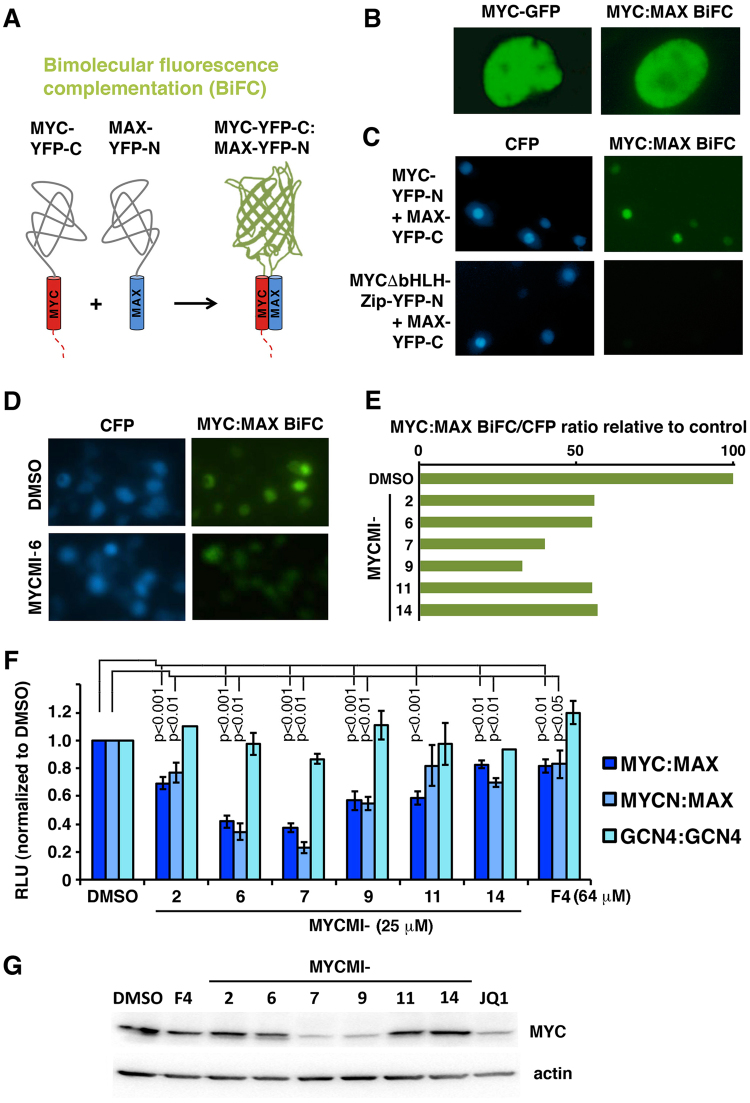
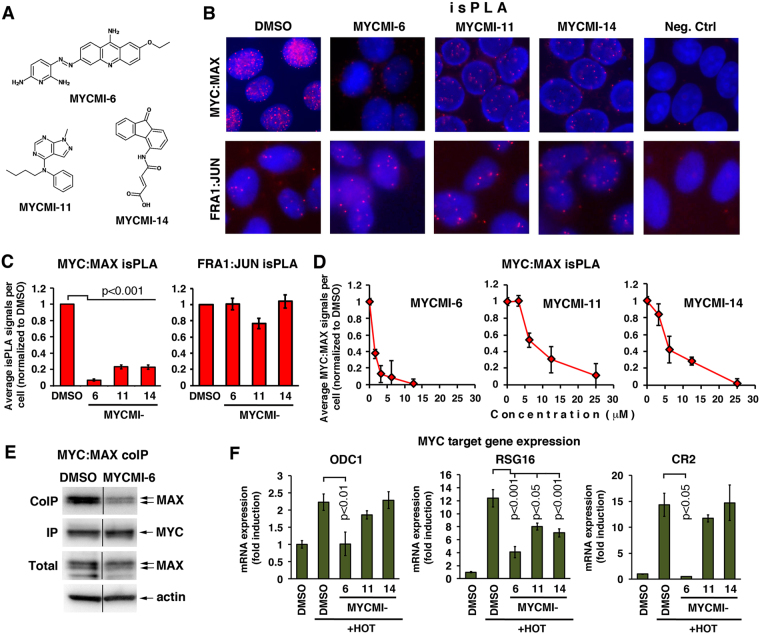
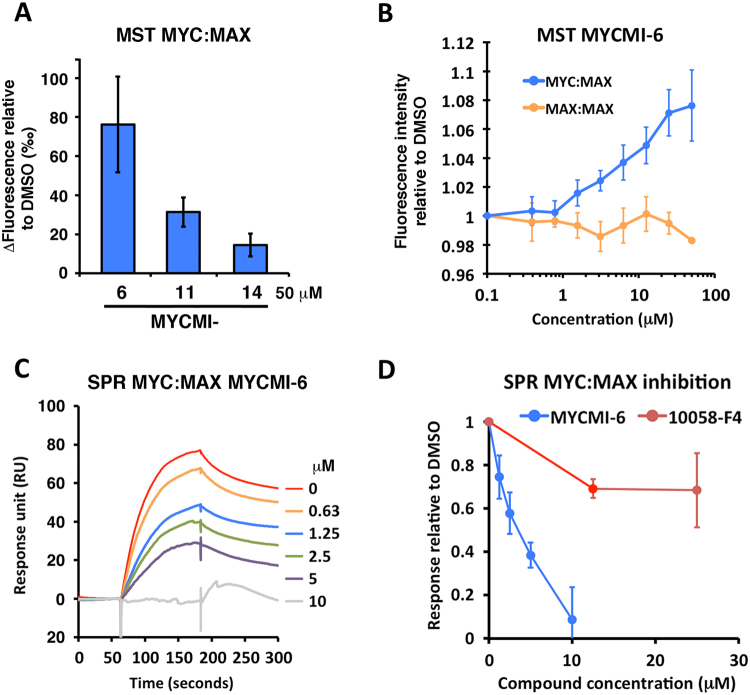
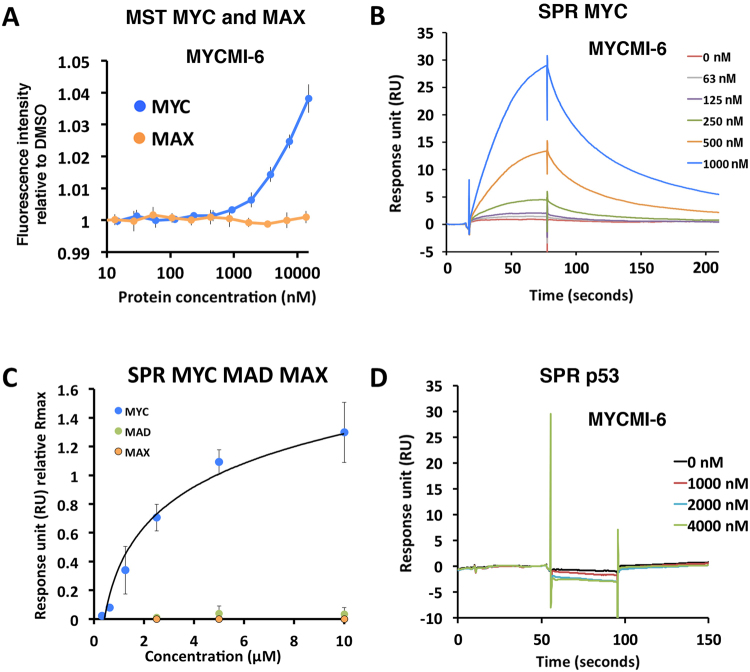
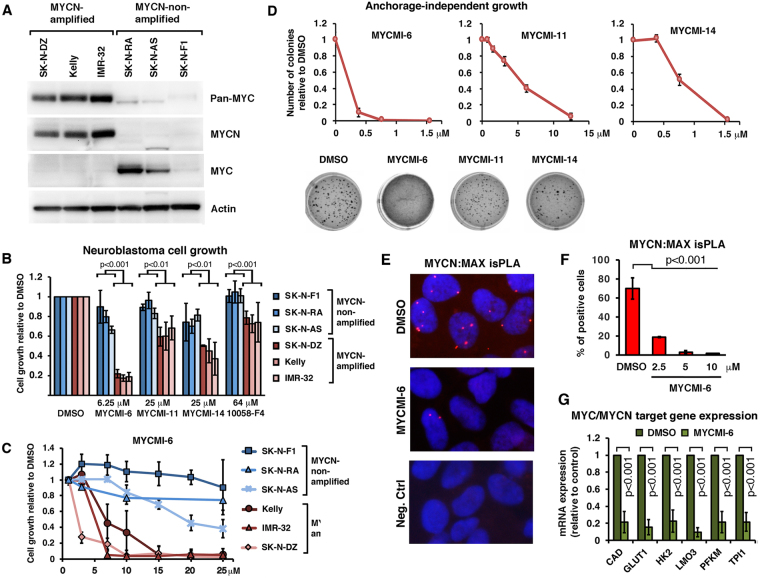
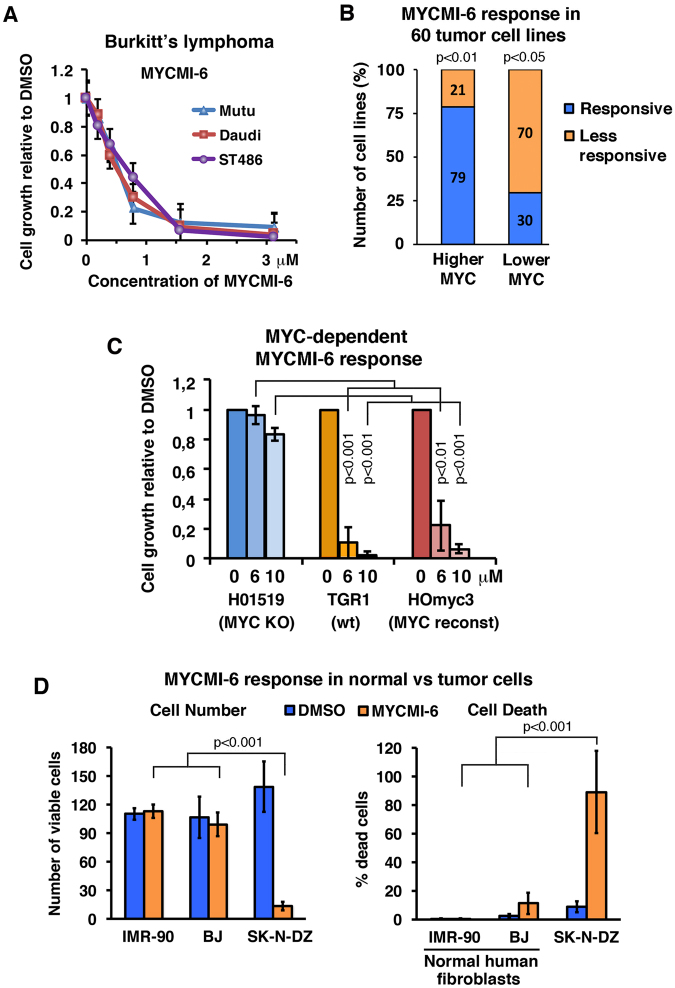
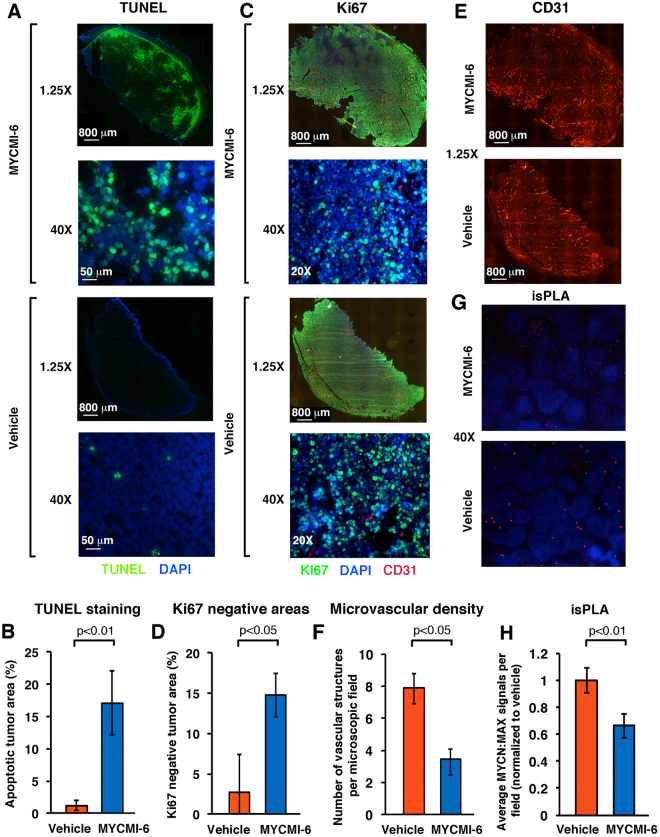
MYC is a key player in tumor development, but unfortunately no specific MYC-targeting drugs are clinically available. MYC is strictly dependent on heterodimerization with MAX for transcription activation. Aiming at targeting this interaction, we identified MYCMI-6 in a cell-based protein interaction screen for small inhibitory molecules. MYCMI-6 exhibits strong selective inhibition of MYC:MAX interaction in cells and in vitro at single-digit micromolar concentrations, as validated by split Gaussia luciferase, in situ proximity ligation, microscale thermophoresis and surface plasmon resonance (SPR) assays. Further, MYCMI-6 blocks MYC-driven transcription and binds selectively to the MYC bHLHZip domain with a KD of 1.6 ± 0.5 μM as demonstrated by SPR. MYCMI-6 inhibits tumor cell growth in a MYC-dependent manner with IC50 concentrations as low as 0.5 μM, while sparing normal cells. The response to MYCMI-6 correlates with MYC expression based on data from 60 human tumor cell lines and is abrogated by MYC depletion. Further, it inhibits MYC:MAX interaction, reduces proliferation and induces massive apoptosis in tumor tissue from a MYC-driven xenograft tumor model without severe side effects. Since MYCMI-6 does not affect MYC expression, it is a unique molecular tool to specifically target MYC:MAX pharmacologically and it has good potential for drug development.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
