

Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling
- PMID: 29972798
- PMCID: PMC6037414
- DOI: 10.1016/j.cmet.2018.06.006
Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling
Abstract
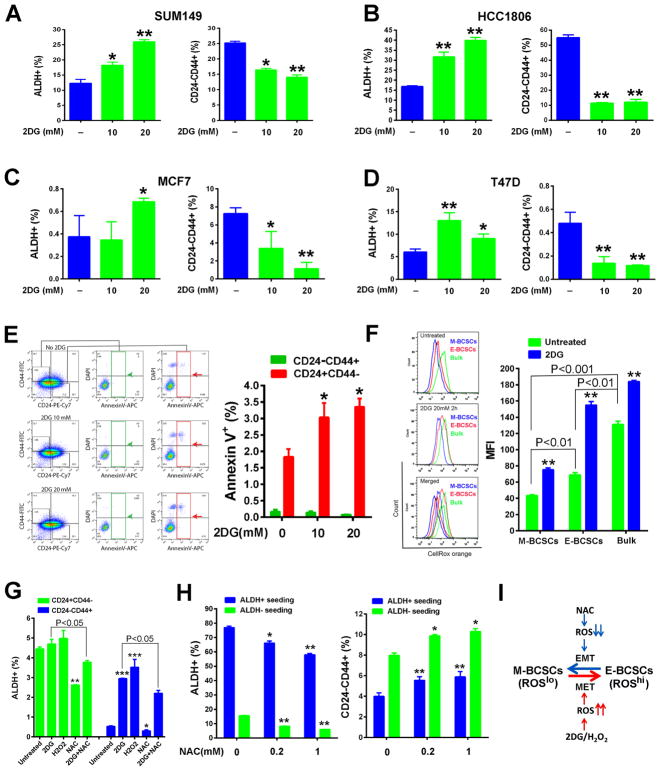
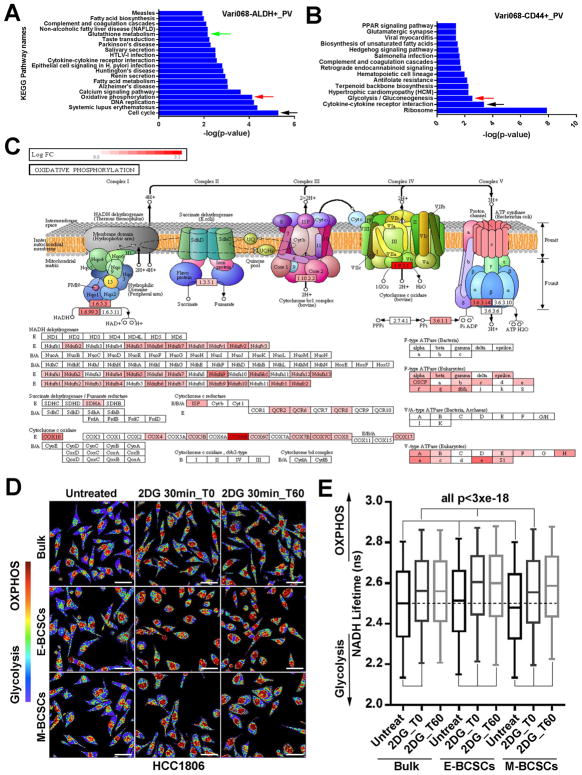
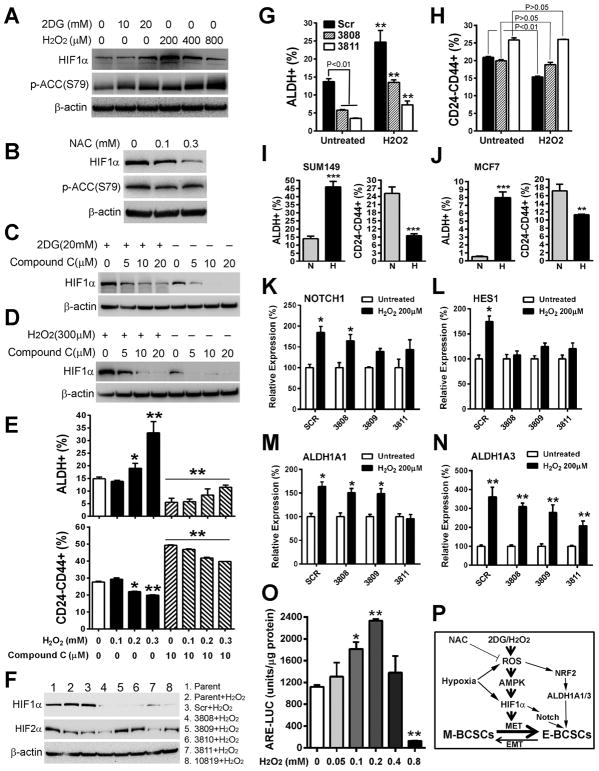
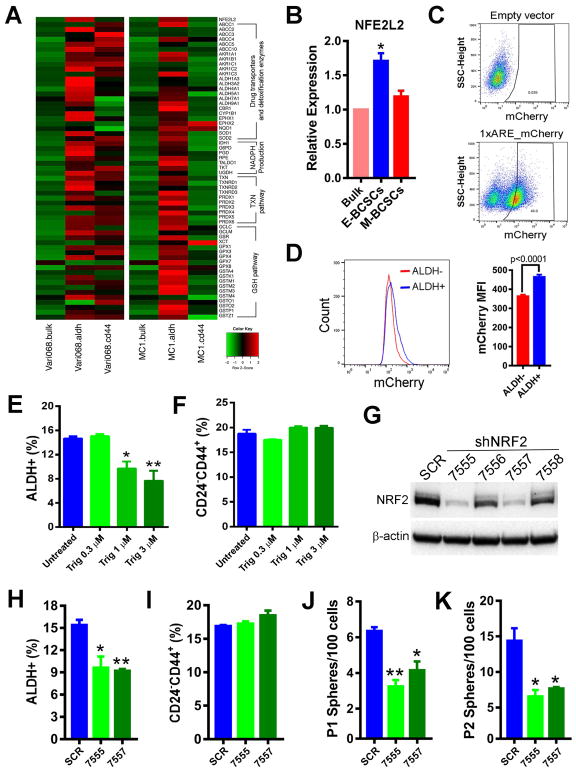
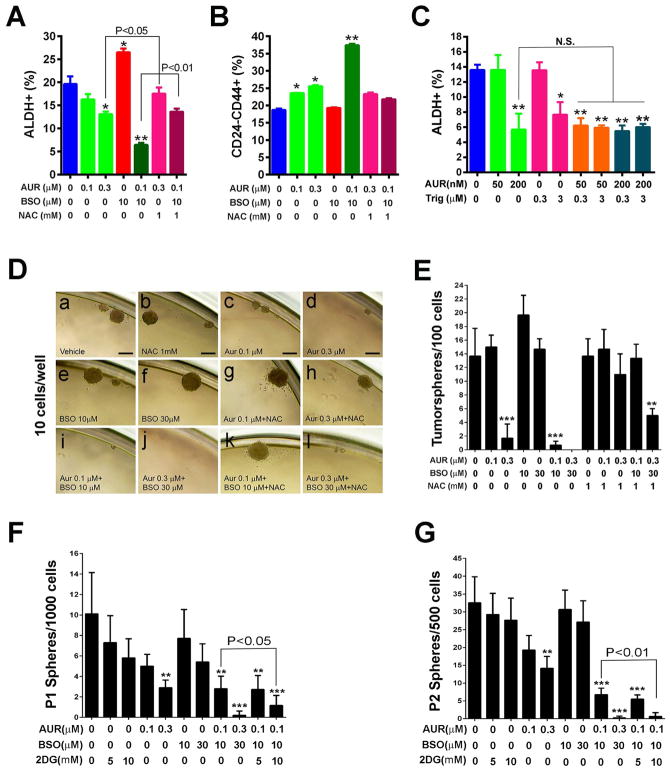
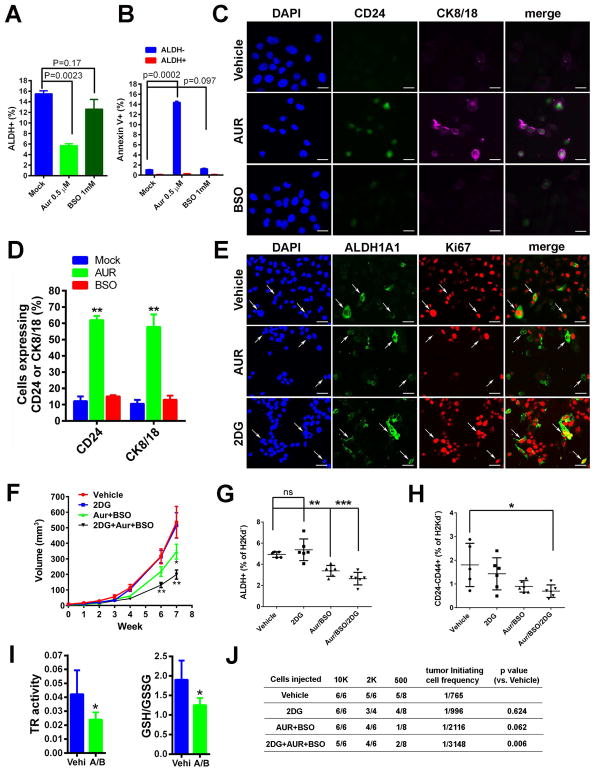
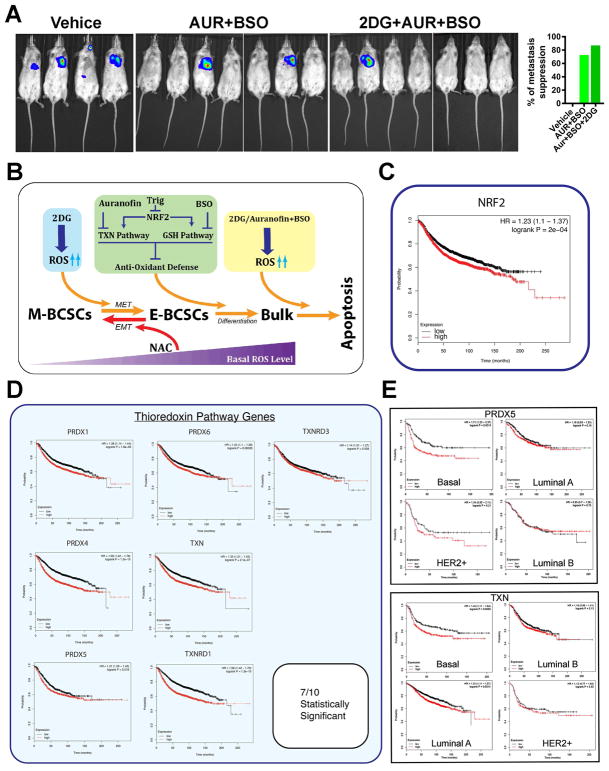
Although breast cancer stem cells (BCSCs) display plasticity transitioning between quiescent mesenchymal-like (M) and proliferative epithelial-like (E) states, how this plasticity is regulated by metabolic or oxidative stress remains poorly understood. Here, we show that M- and E-BCSCs rely on distinct metabolic pathways and display markedly different sensitivities to inhibitors of glycolysis and redox metabolism. Metabolic or oxidative stress generated by 2DG, H2O2, or hypoxia promotes the transition of ROSlo M-BCSCs to a ROShi E-state. This transition is reversed by N-acetylcysteine and mediated by activation of the AMPK-HIF1α axis. Moreover, E-BCSCs exhibit robust NRF2-mediated antioxidant responses, rendering them vulnerable to ROS-induced differentiation and cytotoxicity following suppression of NRF2 or downstream thioredoxin (TXN) and glutathione (GSH) antioxidant pathways. Co-inhibition of glycolysis and TXN and GSH pathways suppresses tumor growth, tumor-initiating potential, and metastasis by eliminating both M- and E-BCSCs. Exploiting metabolic vulnerabilities of distinct BCSC states provides a novel therapeutic approach targeting this critical tumor cell population.
Keywords: HIF1α; NRF2; antioxidant responses; cancer stem cell metabolism; cancer stem cell plasticity; glycolysis; hypoxia; oxidative phosphorylation.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
M.S.W. holds equity in OncoMed Pharmaceuticals.
Figures
References
-
- Arlt A, Sebens S, Krebs S, Geismann C, Grossmann M, Kruse ML, Schreiber S, Schafer H. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene. 2013;32:4825–4835. - PubMed
-
- Balic M, Lin H, Young L, Hawes D, Giuliano A, McNamara G, Datar RH, Cote RJ. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:5615–5621. - PubMed
-
- Brabletz T. To differentiate or not--routes towards metastasis. Nature reviews Cancer. 2012;12:425–436. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
