

Spectrum of genomic variations in Indian patients with progressive familial intrahepatic cholestasis
- PMID: 29973134
- PMCID: PMC6032793
- DOI: 10.1186/s12876-018-0835-6
Spectrum of genomic variations in Indian patients with progressive familial intrahepatic cholestasis
Abstract
Background: Progressive familial intrahepatic cholestasis (PFIC) is caused by variations in ATP8B1, ABCB11 or ABCB4 genes. Data on genetic variations in Indian patients with PFIC are lacking.
Methods: Coding and splice regions of the three genes were sequenced in unrelated Indian children with PFIC phenotype. The variations identified were looked for in parents, 30 healthy persons and several variation databases, and their effect was assessed in-silico.
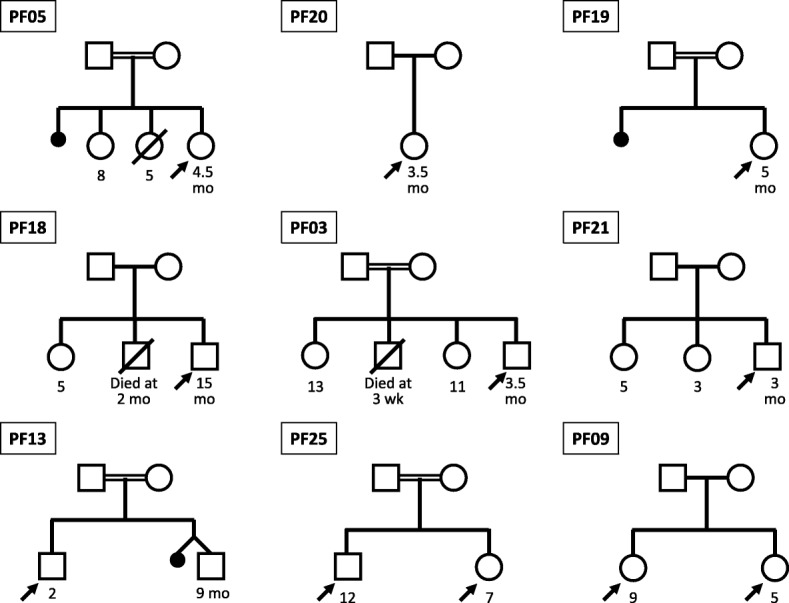
Results: Among 25 children (aged 1-144 months), nine (36%) had unique major genomic variations (ATP8B1: 4, ABCB11: 3 and ABCB4: 2). Seven had homozygous variations, which were assessed as 'pathogenic' or 'likely pathogenic'. These included: (i) four amino acid substitutions (ATP8B1: c.1660G > A/p.Asp554Asn and c.2941G > A/p.Glu981Lys; ABCB11: c.548 T > C/p.Met183Thr; ABCB4: c.431G > A/p.Arg144Gln); (ii) one 3-nucleotide deletion causing an amino acid deletion (ATP8B1: c.1587_1589delCTT/p.Phe529del); (iii) one single-nucleotide deletion leading to frame-shift and premature termination (ABCB11: c.1360delG/p.Val454Ter); and (iv) a complex inversion of 4 nucleotides with a single-nucleotide insertion leading to frame-shift and premature termination (ATP8B1: c.[589_592inv;592_593insA]/p.Gly197LeufsTer10). Two variations were found in heterozygous form: (i) a splice-site variation likely to cause abnormal splicing (ABCB11: c.784 + 1G > C), and (ii) a nucleotide substitution that created a premature stop codon (ABCB4: c.475C > T/p.Arg159Ter); these were considered as variations of uncertain significance. Three of the nine variations were novel.
Conclusions: Nine major genomic variations, including three novel ones, were identified in nearly one-third of Indian children with PFIC. No variation was identified in nearly two-thirds of patients, who may have been related to variations in promoter or intronic regions of the three PFIC genes, or in other bile-salt transport genes.
Keywords: Genetic variation; Hereditary cholestasis; Polymorphism; Progressive familial intrahepatic cholestasis.
Conflict of interest statement
Ethics approval and consent to participate
The study was approved by Ethics Committee of the Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, India. An informed written consent was obtained from one of the parents for each child studied.
Consent for publication
The consent document for study subjects included consent for publication of data (after removing any identifying information).
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
Heterozygous mutations of ATP8B1, ABCB11 and ABCB4 cause mild forms of Progressive Familial Intrahepatic Cholestasis in a pediatric cohort.Gastroenterol Hepatol. 2022 Oct;45(8):585-592. doi: 10.1016/j.gastrohep.2021.12.005. Epub 2021 Dec 20. Gastroenterol Hepatol. 2022. PMID: 34942279 English, Spanish.
-
ATP8B1, ABCB11, and ABCB4 Genes Defects: Novel Mutations Associated with Cholestasis with Different Phenotypes and Outcomes.J Pediatr. 2021 Sep;236:113-123.e2. doi: 10.1016/j.jpeds.2021.04.040. Epub 2021 Apr 27. J Pediatr. 2021. PMID: 33915153
-
The spectrum of novel ABCB11 gene variations in children with progressive familial intrahepatic cholestasis type 2 in Pakistani cohorts.Sci Rep. 2024 Aug 14;14(1):18876. doi: 10.1038/s41598-024-59945-0. Sci Rep. 2024. PMID: 39143102 Free PMC article.
-
Molecular overview of progressive familial intrahepatic cholestasis.World J Gastroenterol. 2020 Dec 21;26(47):7470-7484. doi: 10.3748/wjg.v26.i47.7470. World J Gastroenterol. 2020. PMID: 33384548 Free PMC article. Review.
-
Autoimmune BSEP disease: disease recurrence after liver transplantation for progressive familial intrahepatic cholestasis.Clin Rev Allergy Immunol. 2015 Jun;48(2-3):273-84. doi: 10.1007/s12016-014-8457-4. Clin Rev Allergy Immunol. 2015. PMID: 25342496 Review.
Cited by
-
Phenotypic and Molecular Characteristics of Children with Progressive Familial Intrahepatic Cholestasis in South China.Pediatr Gastroenterol Hepatol Nutr. 2020 Nov;23(6):558-566. doi: 10.5223/pghn.2020.23.6.558. Epub 2020 Nov 5. Pediatr Gastroenterol Hepatol Nutr. 2020. PMID: 33215027 Free PMC article.
-
Progressive Familial Intrahepatic Cholestasis: Need for Genetic Analysis Before Liver Transplantation.J Clin Exp Hepatol. 2022 Mar-Apr;12(2):686-688. doi: 10.1016/j.jceh.2021.06.009. Epub 2021 Jun 18. J Clin Exp Hepatol. 2022. PMID: 35535065 Free PMC article.
-
Genetic Variants and Long-Term Outcomes in Korean Children with Progressive Familial Intrahepatic Cholestasis.Pediatr Gastroenterol Hepatol Nutr. 2025 Jul;28(4):245-255. doi: 10.5223/pghn.2025.28.4.245. Epub 2025 Jul 7. Pediatr Gastroenterol Hepatol Nutr. 2025. PMID: 40697762 Free PMC article.
-
Clinical and molecular genetic characteristics of pediatric PFIC3 patients: three novel variants and prognosis for parental liver transplantation.Orphanet J Rare Dis. 2025 Apr 8;20(1):164. doi: 10.1186/s13023-025-03670-y. Orphanet J Rare Dis. 2025. PMID: 40200381 Free PMC article.
-
Rare variant contribution to cholestatic liver disease in a South Asian population in the United Kingdom.Sci Rep. 2023 May 19;13(1):8120. doi: 10.1038/s41598-023-33391-w. Sci Rep. 2023. PMID: 37208429 Free PMC article.
References
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
