

MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma
- PMID: 29973406
- PMCID: PMC8054766
- DOI: 10.1126/scitranslmed.aan4470
MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma
Abstract
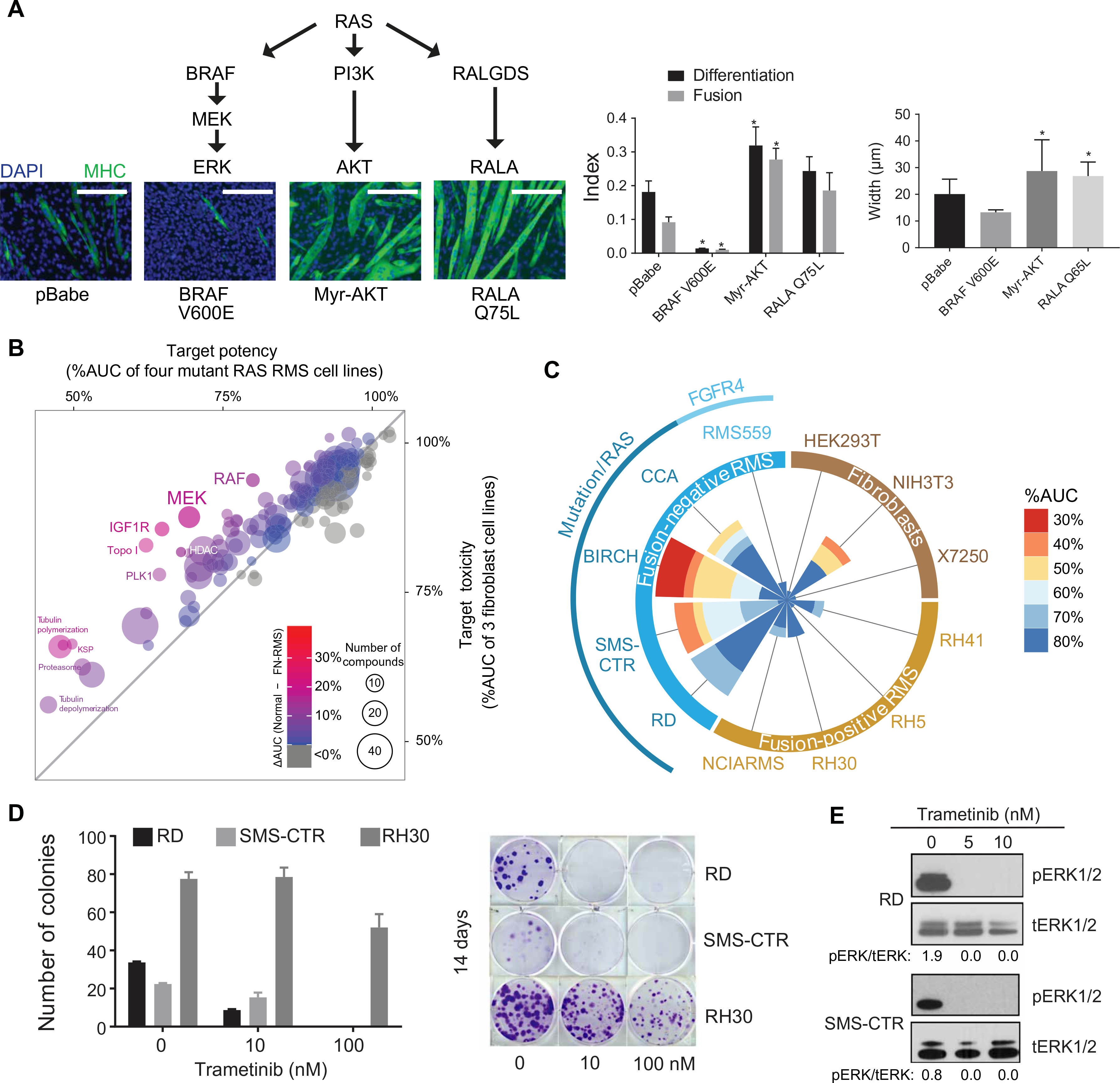
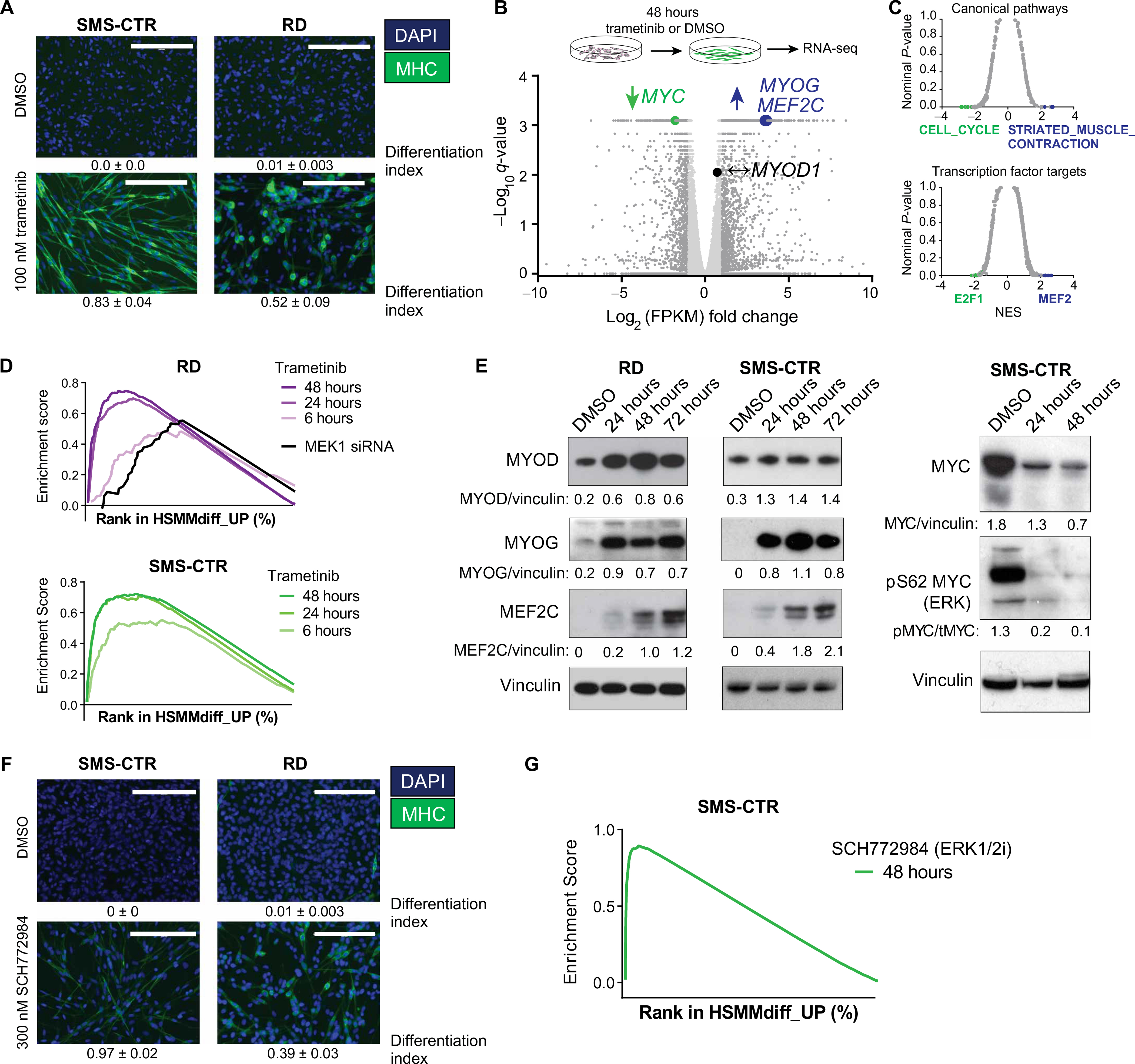
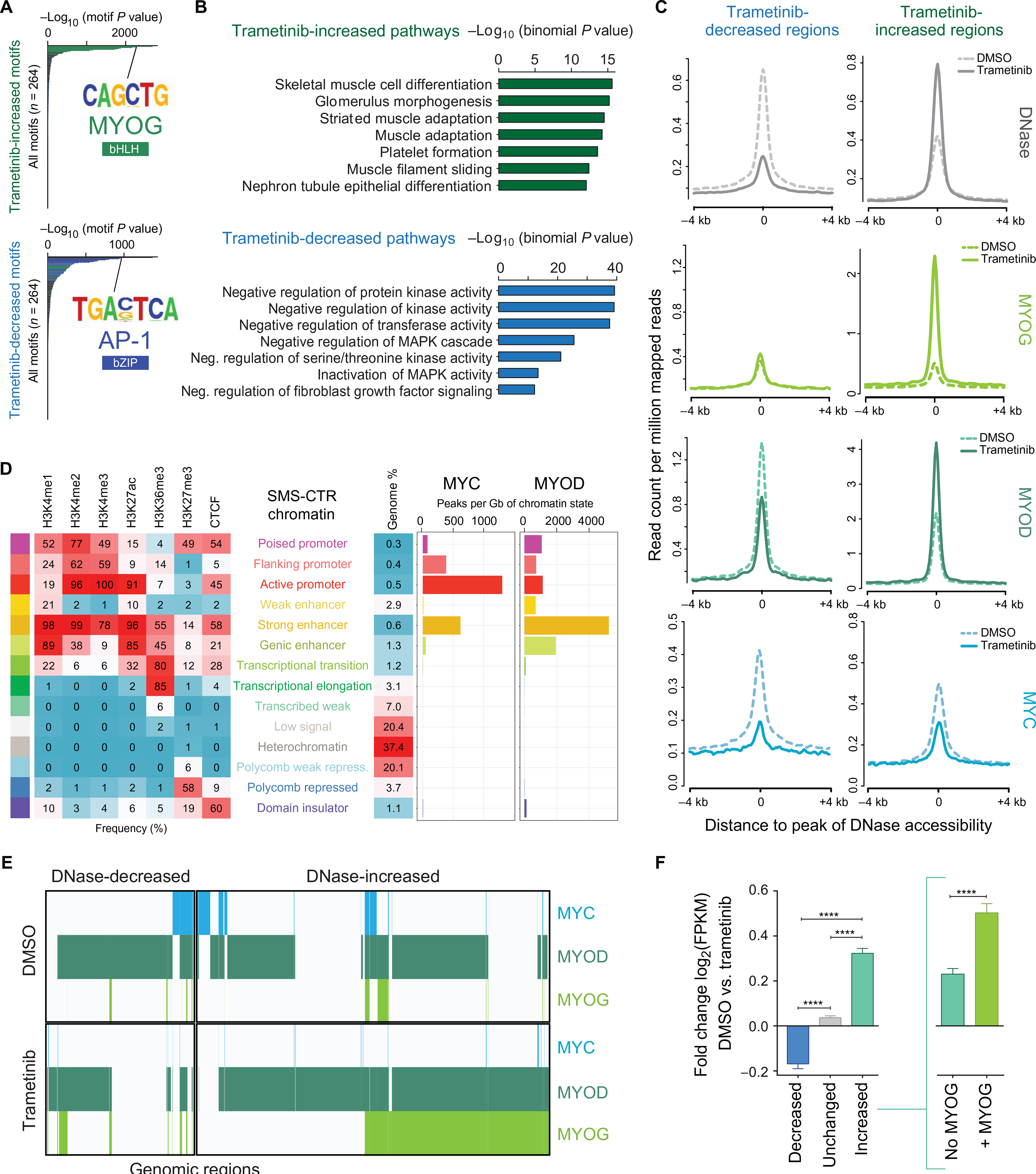
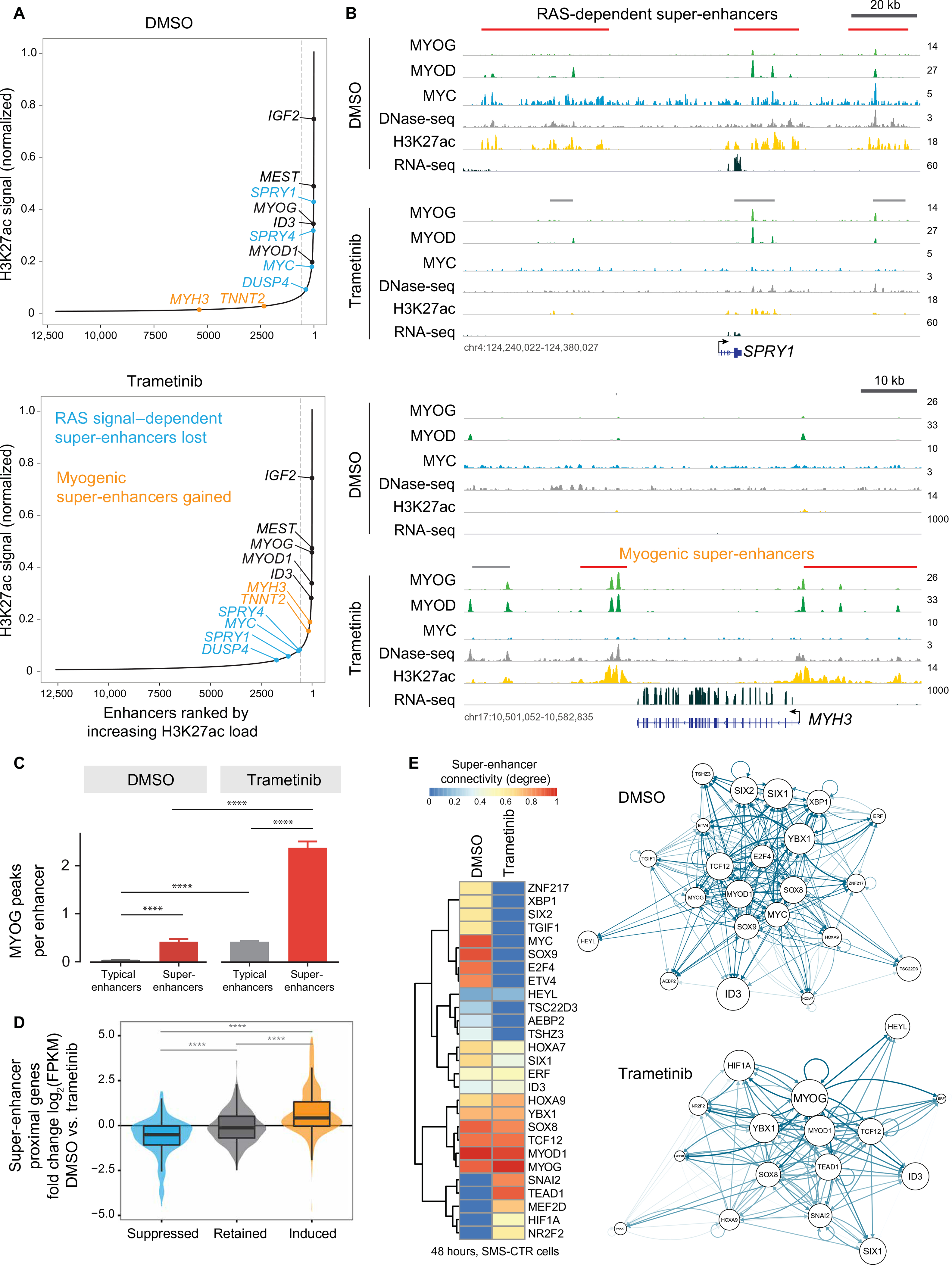
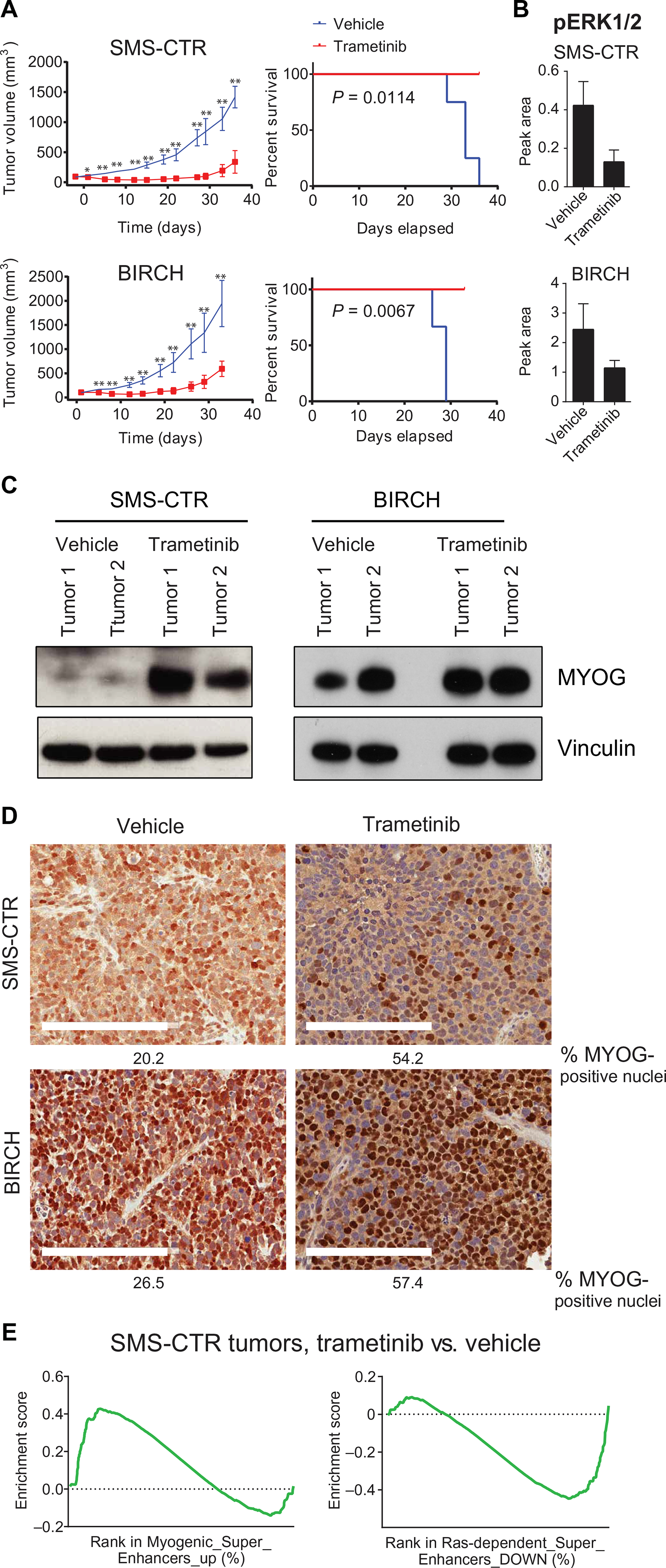
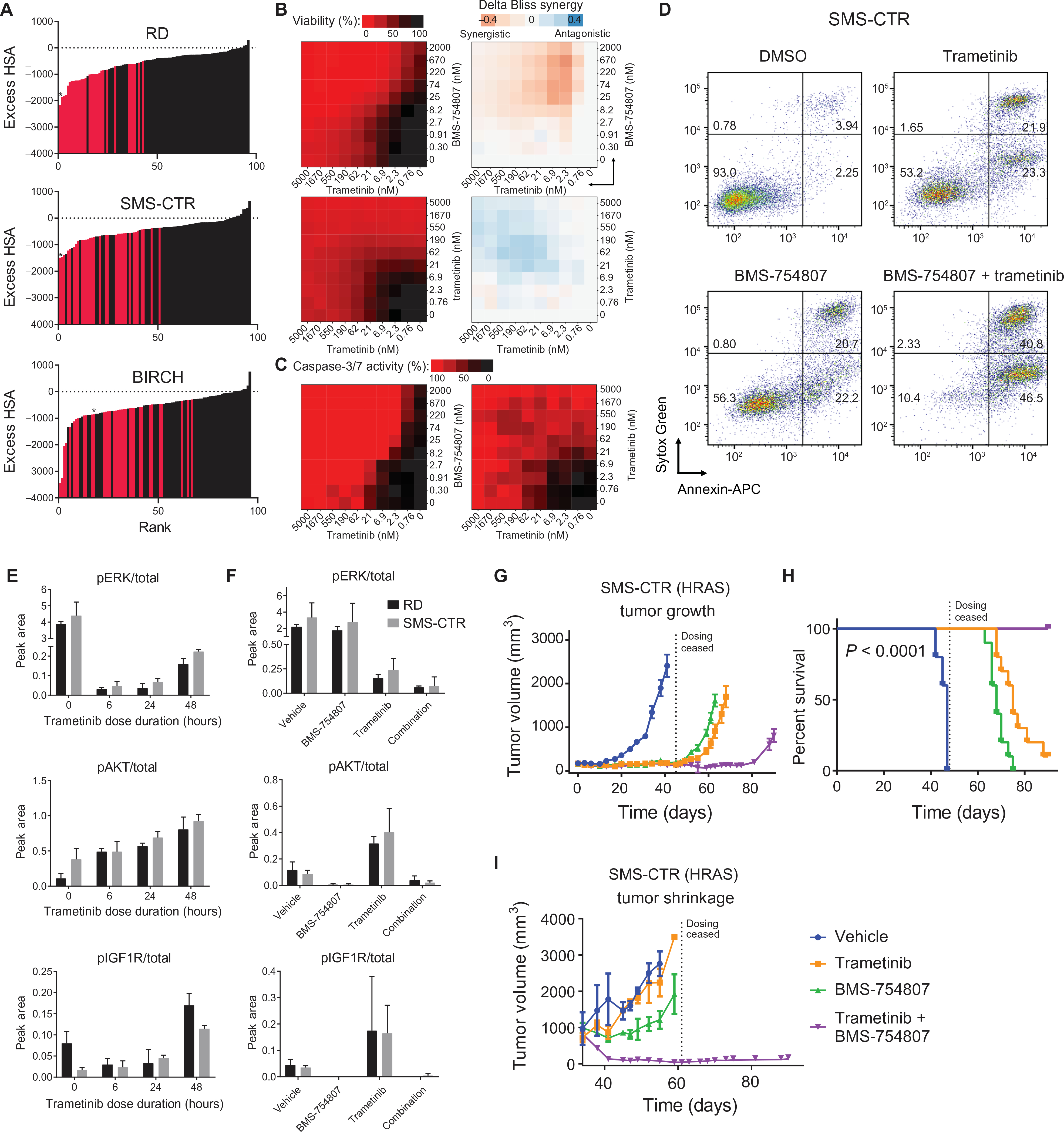
The RAS isoforms are frequently mutated in many types of human cancers, including PAX3/PAX7 fusion-negative rhabdomyosarcoma. Pediatric RMS arises from skeletal muscle progenitor cells that have failed to differentiate normally. The role of mutant RAS in this differentiation blockade is incompletely understood. We demonstrate that oncogenic RAS, acting through the RAF-MEK [mitogen-activated protein kinase (MAPK) kinase]-ERK (extracellular signal-regulated kinase) MAPK effector pathway, inhibits myogenic differentiation in rhabdomyosarcoma by repressing the expression of the prodifferentiation myogenic transcription factor, MYOG. This repression is mediated by ERK2-dependent promoter-proximal stalling of RNA polymerase II at the MYOG locus. Small-molecule screening with a library of mechanistically defined inhibitors showed that RAS-driven RMS is vulnerable to MEK inhibition. MEK inhibition with trametinib leads to the loss of ERK2 at the MYOG promoter and releases the transcriptional stalling of MYOG expression. MYOG subsequently opens chromatin and establishes super-enhancers at genes required for late myogenic differentiation. Furthermore, trametinib, in combination with an inhibitor of IGF1R, potently decreases rhabdomyosarcoma cell viability and slows tumor growth in xenograft models. Therefore, this combination represents a potential therapeutic for RAS-mutated rhabdomyosarcoma.
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures

References
-
- Sos ML, Fischer S, Ullrich R, Peifer M, Heuckmann JM, Koker M, Heynck S, Stückrath I, Weiss J, Fischer F, Michel K, Goel A, Regales L, Politi KA, Perera S, Getlik M, Heukamp LC, Ansén S, Zander T, Beroukhim R, Kashkar H, Shokat KM, Sellers WR, Rauh D, Orr C, Hoeflich KP, Friedman L, Wong K-K, Pao W, Thomas RK, Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc. Natl. Acad. Sci. U.S.A 106, 18351–18356 (2009). - PMC - PubMed
-
- Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen H-Y, Li B, Swoboda RK, Wilson M, Vultur A, Fukunaba-Kalabis M, Wubbenhorst B, Chen TY, Liu Q, Sproesser K, DeMarini DJ, Gilmer TM, Martin A-M, Marmorstein R, Schultz DC, Speicher DW, Karakousis GC, Xu W, Amaravadi RK, Xu X, Schuchter LM, Herlyn M, Nathanson KL, Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep 4, 1090–1099 (2013). - PMC - PubMed
-
- Darley RL, Burnett AK, Mutant RAS inhibits neutrophil but not macrophage differentiation and allows continued growth of neutrophil precursors. Exp. Hematol 27, 1599–1608 (1999). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
