

SMAD4 Prevents Flow Induced Arteriovenous Malformations by Inhibiting Casein Kinase 2
- PMID: 29976569
- PMCID: PMC6309254
- DOI: 10.1161/CIRCULATIONAHA.118.033842
SMAD4 Prevents Flow Induced Arteriovenous Malformations by Inhibiting Casein Kinase 2
Abstract
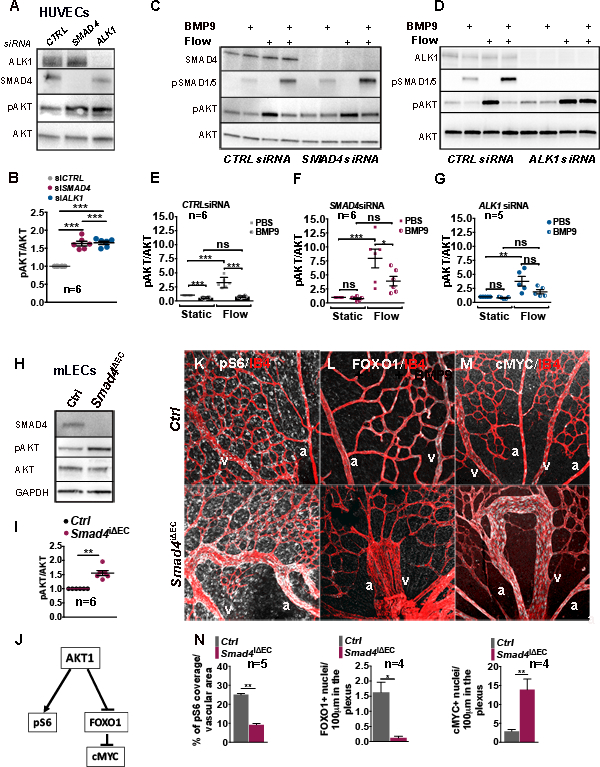
Background: Hereditary hemorrhagic telangiectasia (HHT) is an inherited vascular disorder that causes arteriovenous malformations (AVMs). Mutations in the genes encoding Endoglin ( ENG) and activin-receptor-like kinase 1 ( AVCRL1 encoding ALK1) cause HHT type 1 and 2, respectively. Mutations in the SMAD4 gene are present in families with juvenile polyposis-HHT syndrome that involves AVMs. SMAD4 is a downstream effector of transforming growth factor-β (TGFβ)/bone morphogenetic protein (BMP) family ligands that signal via activin-like kinase receptors (ALKs). Ligand-neutralizing antibodies or inducible, endothelial-specific Alk1 deletion induce AVMs in mouse models as a result of increased PI3K (phosphatidylinositol 3-kinase)/AKT (protein kinase B) signaling. Here we addressed if SMAD4 was required for BMP9-ALK1 effects on PI3K/AKT pathway activation.
Methods: The authors generated tamoxifen-inducible, postnatal, endothelial-specific Smad4 mutant mice ( Smad4iΔEC).
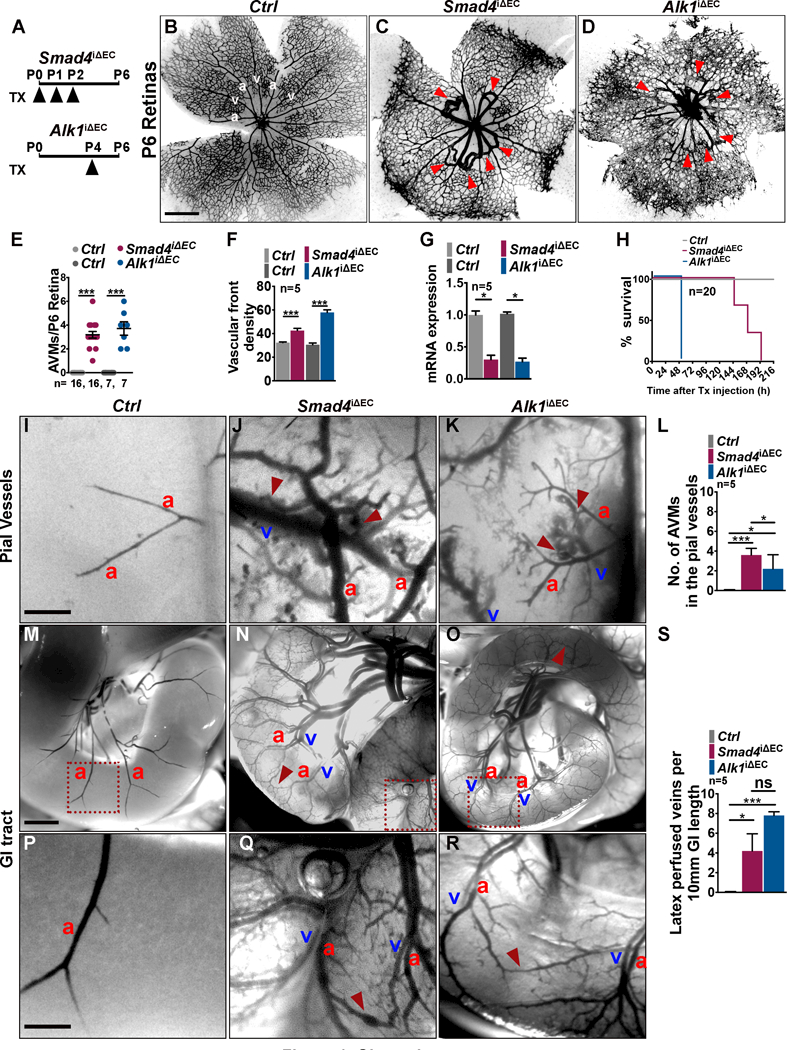
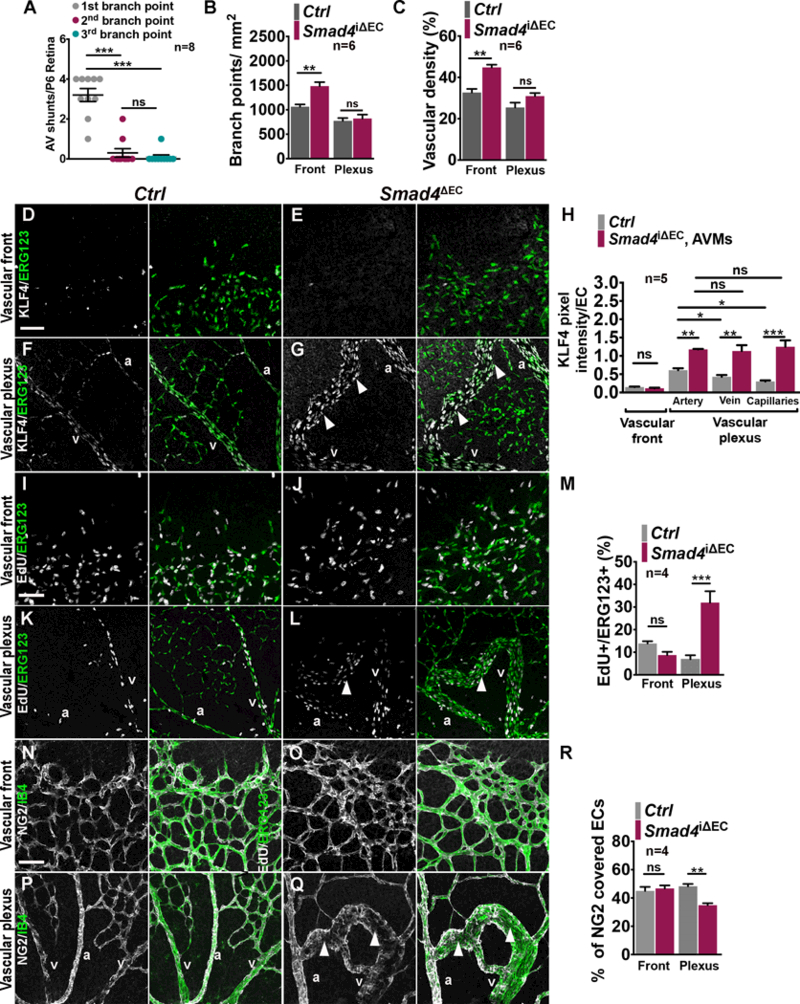
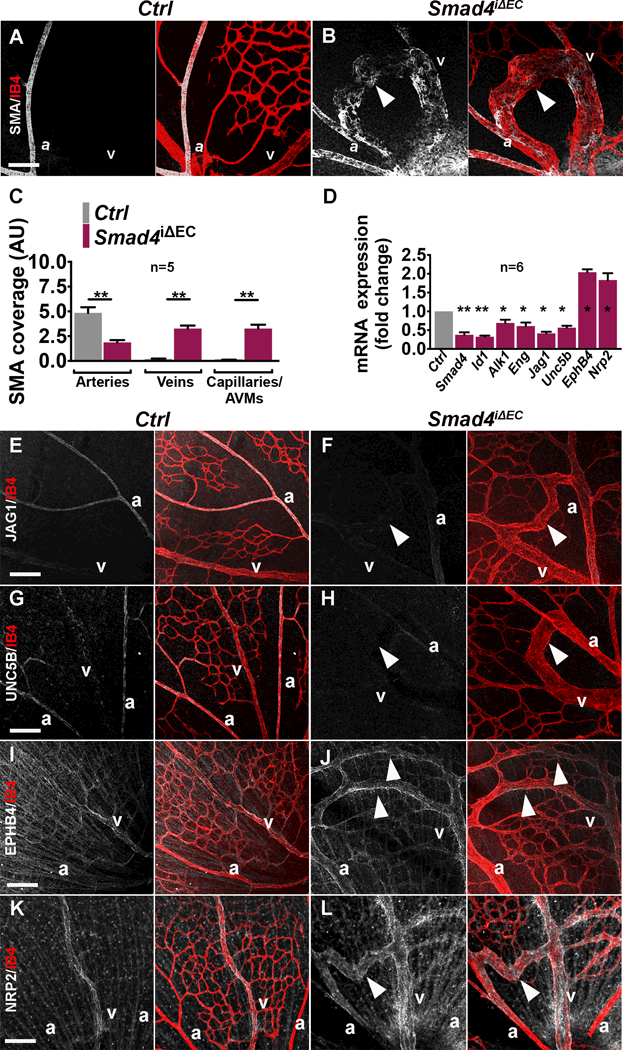
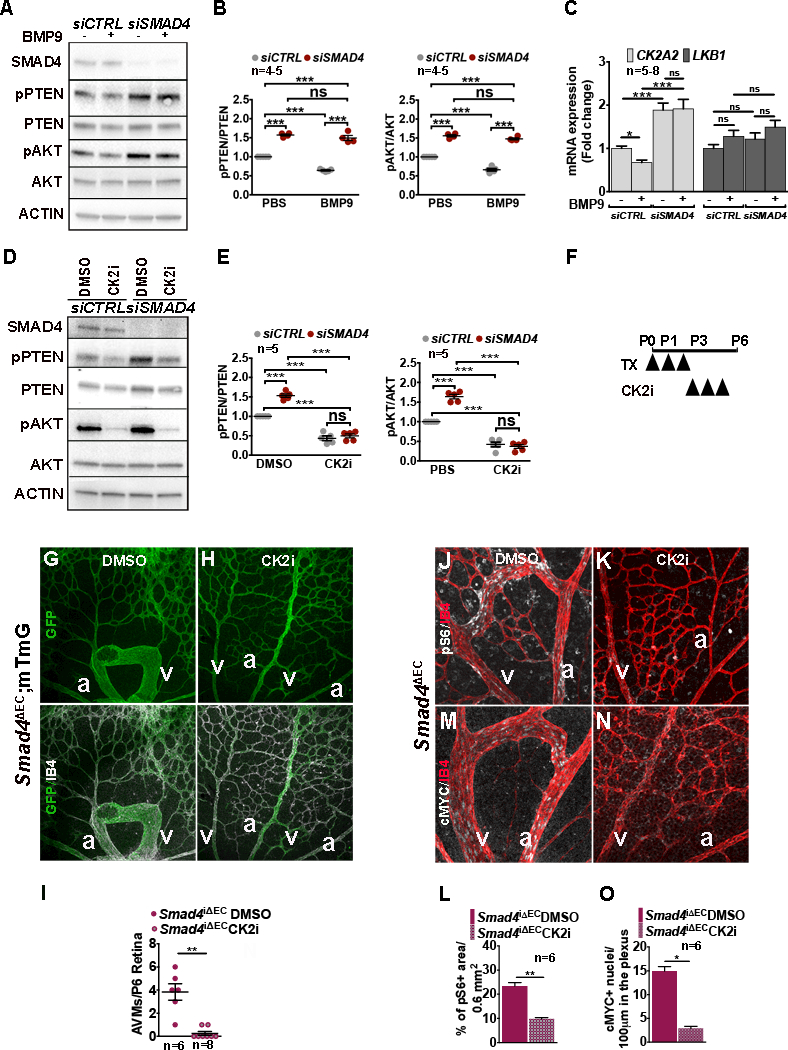
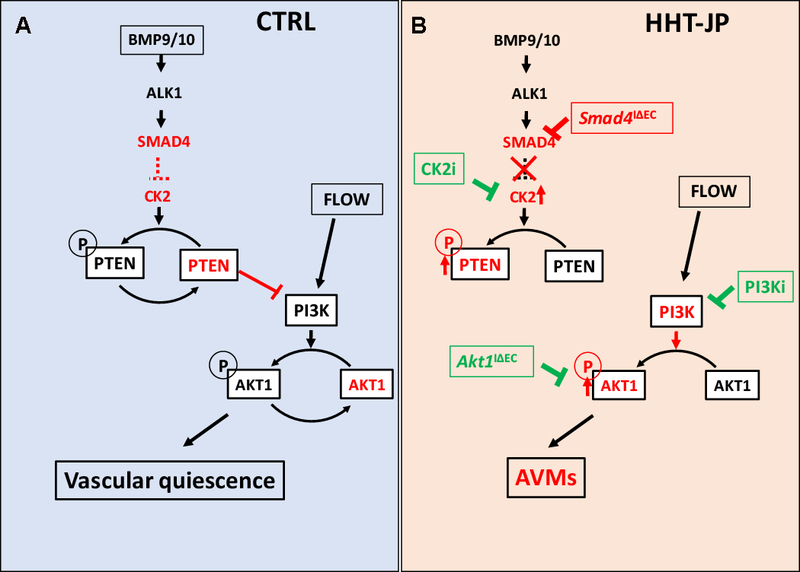
Results: We found that loss of endothelial Smad4 resulted in AVM formation and lethality. AVMs formed in regions with high blood flow in developing retinas and other tissues. Mechanistically, BMP9 signaling antagonized flow-induced AKT activation in an ALK1- and SMAD4-dependent manner. Smad4iΔEC endothelial cells in AVMs displayed increased PI3K/AKT signaling, and pharmacological PI3K inhibitors or endothelial Akt1 deletion both rescued AVM formation in Smad4iΔEC mice. BMP9-induced SMAD4 inhibited casein kinase 2 ( CK2) transcription, in turn limiting PTEN phosphorylation and AKT activation. Consequently, CK2 inhibition prevented AVM formation in Smad4iΔEC mice.
Conclusions: Our study reveals SMAD4 as an essential effector of BMP9-10/ALK1 signaling that affects AVM pathogenesis via regulation of CK2 expression and PI3K/AKT1 activation.
Keywords: Telangiectasia, hereditary hemorrhagic; activin receptors, type II; arteriovenous malformations; casein kinase; phosphatidylinositol 3-kinase; proto-oncogene proteins c-akt.
Conflict of interest statement
Conflict of Interest Disclosures
None.
Figures

References
-
- Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev 2010;24:203–219. - PubMed
-
- Gallione CJ, Klaus DJ, Yeh EY, Stenzel TT, Xue Y, Anthony KB, McAllister KA, Baldwin MA, Berg JN, Lux A, Smith JD, Vary CP, Craigen WJ, Westermann CJ, Warner ML, Miller YE, Jackson CE, Guttmacher AE and Marchuk DA. Mutation and expression analysis of the endoglin gene in hereditary hemorrhagic telangiectasia reveals null alleles. Hum Mutat 1998;11:286–294. - PubMed
-
- Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, Guttmacher AE, Jackson CE, Attisano L, Kucherlapati R, Porteous ME and Marchuk DA. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet 1996;13:189–195. - PubMed
-
- McDonald J, Bayrak-Toydemir P and Pyeritz RE. Hereditary hemorrhagic telangiectasia: an overview of diagnosis, management, and pathogenesis. Genet Med 2011;13:607–616. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
