

A network of epigenomic and transcriptional cooperation encompassing an epigenomic master regulator in cancer
- PMID: 29977600
- PMCID: PMC6026491
- DOI: 10.1038/s41540-018-0061-4
A network of epigenomic and transcriptional cooperation encompassing an epigenomic master regulator in cancer
Abstract
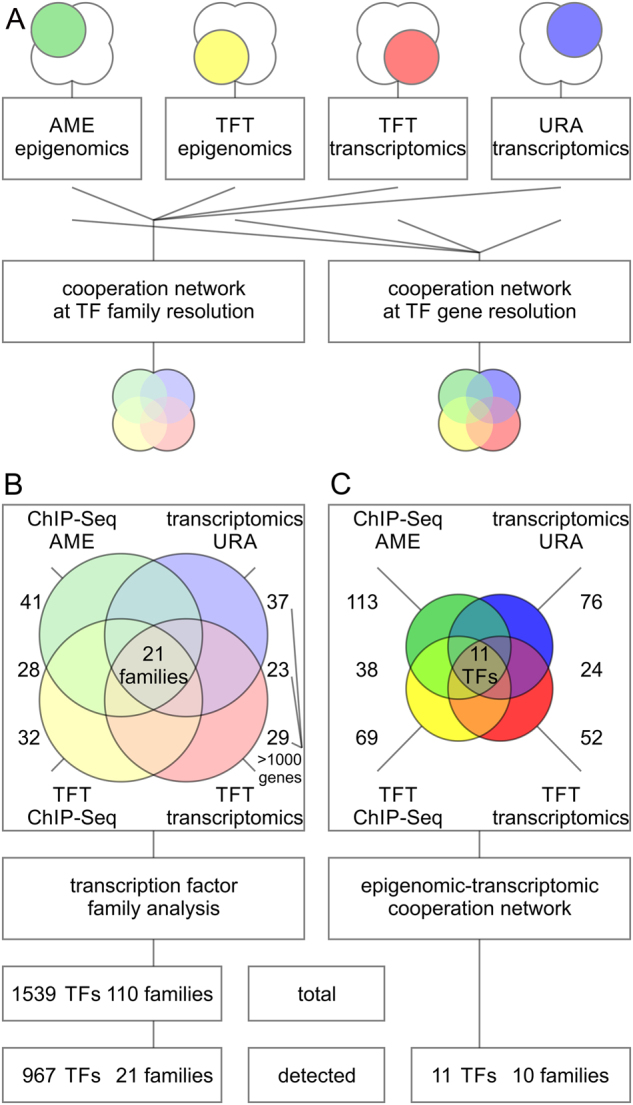
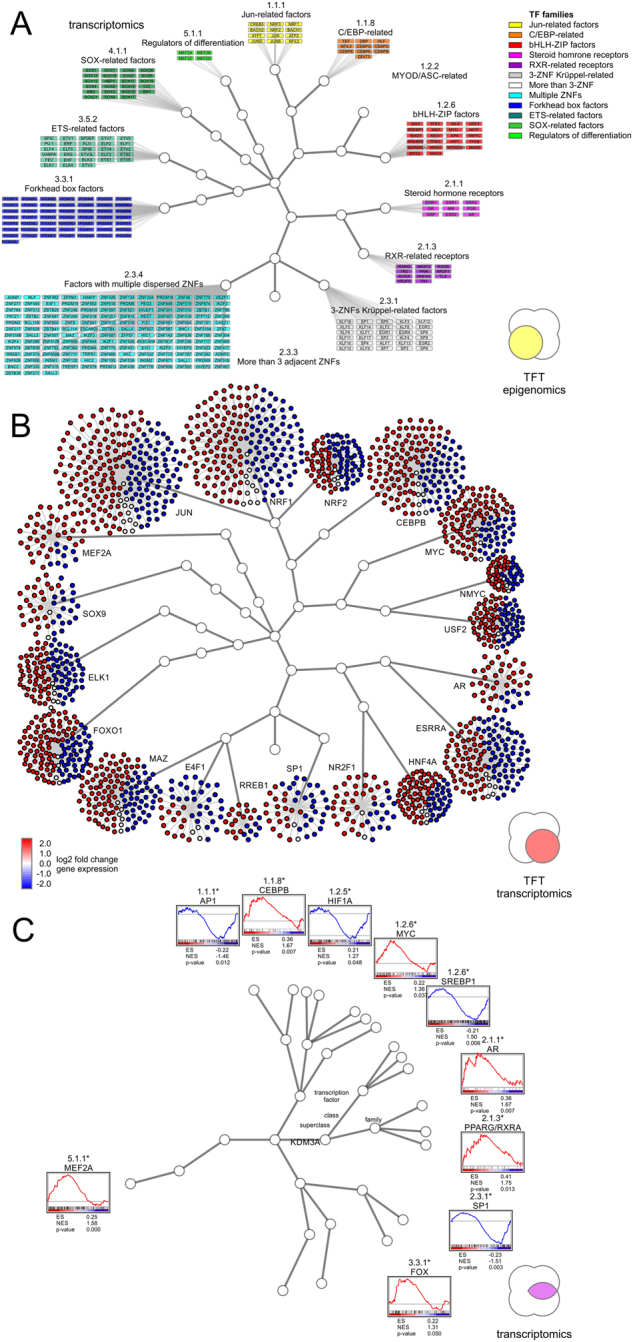
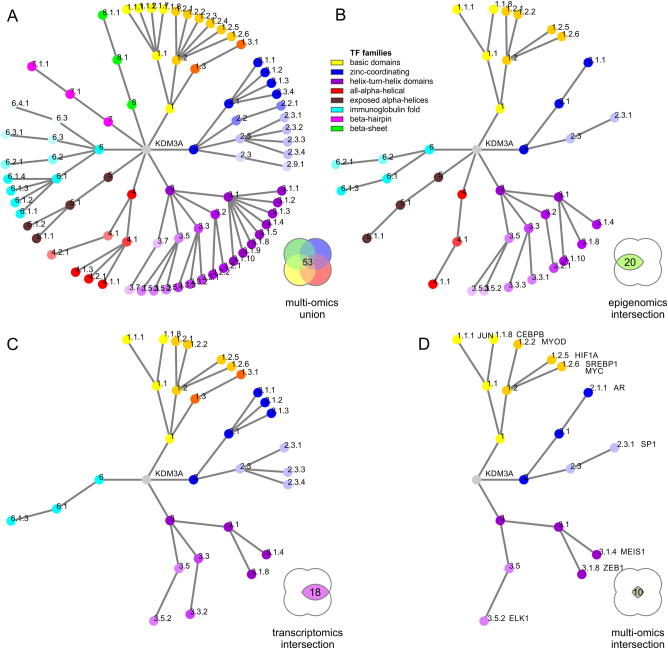
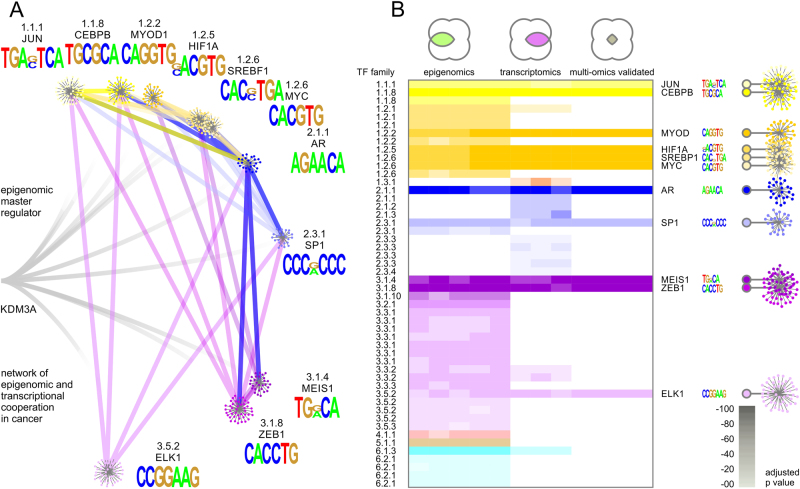
Coordinated experiments focused on transcriptional responses and chromatin states are well-equipped to capture different epigenomic and transcriptomic levels governing the circuitry of a regulatory network. We propose a workflow for the genome-wide identification of epigenomic and transcriptional cooperation to elucidate transcriptional networks in cancer. Gene promoter annotation in combination with network analysis and sequence-resolution of enriched transcriptional motifs in epigenomic data reveals transcription factor families that act synergistically with epigenomic master regulators. By investigating complementary omics levels, a close teamwork of the transcriptional and epigenomic machinery was discovered. The discovered network is tightly connected and surrounds the histone lysine demethylase KDM3A, basic helix-loop-helix factors MYC, HIF1A, and SREBF1, as well as differentiation factors AP1, MYOD1, SP1, MEIS1, ZEB1, and ELK1. In such a cooperative network, one component opens the chromatin, another one recognizes gene-specific DNA motifs, others scaffold between histones, cofactors, and the transcriptional complex. In cancer, due to the ability to team up with transcription factors, epigenetic factors concert mitogenic and metabolic gene networks, claiming the role of a cancer master regulators or epioncogenes. Significantly, specific histone modification patterns are commonly associated with open or closed chromatin states, and are linked to distinct biological outcomes by transcriptional activation or repression. Disruption of patterns of histone modifications is associated with the loss of proliferative control and cancer. There is tremendous therapeutic potential in understanding and targeting histone modification pathways. Thus, investigating cooperation of chromatin remodelers and the transcriptional machinery is not only important for elucidating fundamental mechanisms of chromatin regulation, but also necessary for the design of targeted therapeutics.
Conflict of interest statement
The authors declare no competing interests.All experimental protocols were approved by the Institutional Review Boards at the University of California Merced. The study was carried out as part of IRB UCM13-0025 of the University of California Merced and as part of dbGap ID 5094 for study accession phs000178.v9.p8 on somatic mutations in cancer and conducted in accordance with the Helsinki Declaration of 1975.
Figures

References
-
- Filipp FV. Epioncogenes in cancer—identification of epigenomic and transcriptomic cooperation networks by multi-omics integration of ChIP-Seq and RNA-Seq data. Syst. Biol. Methods Mol. Biol. 2019;1800:101–121.
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
