

Clinical data and genetic mutation in Kallmann syndrome with CHARGE syndrome: Case report and pedigree analysis
- PMID: 29979396
- PMCID: PMC6076089
- DOI: 10.1097/MD.0000000000011284
Clinical data and genetic mutation in Kallmann syndrome with CHARGE syndrome: Case report and pedigree analysis
Abstract
Rationale: This study aimed to investigate the genetic mutation characteristics of Kallmann syndrome (KS) with CHARGE syndrome through the clinical features and genetic analysis of a pediatric patient with KS in one pedigree.
Patient concerns: Developmental disorders with olfactory abnormalities, developmental lag, heart malformations, external genital malformations.
Diagnoses: KS combined with some clinical characteristics of CHARGE syndrome. Molecular genetic analysis found that mutation occurred in the CHD7 gene.
Interventions: One pediatric patient's clinical data were collected and genomic DNA extracted from the peripheral blood. Nextgeneration gene sequencing technology was used to detect pathogenic genes, and the Sanger method was applied to perform pedigree verification for the detected suspicious pathogenic mutations.
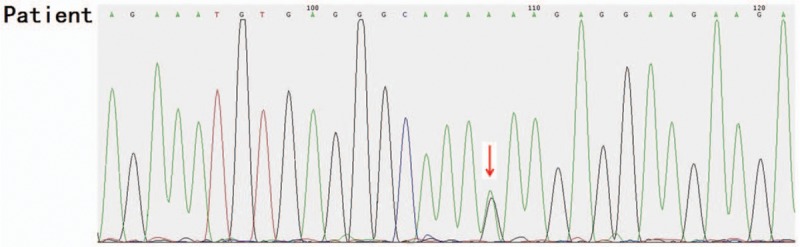
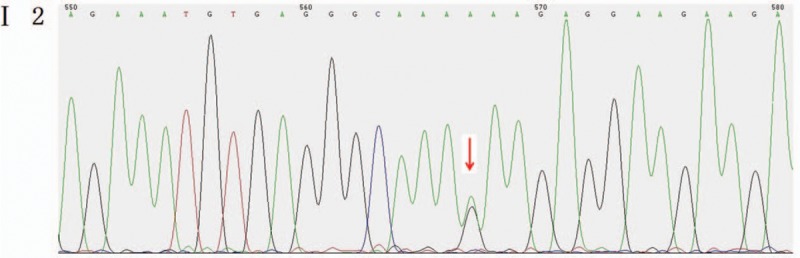
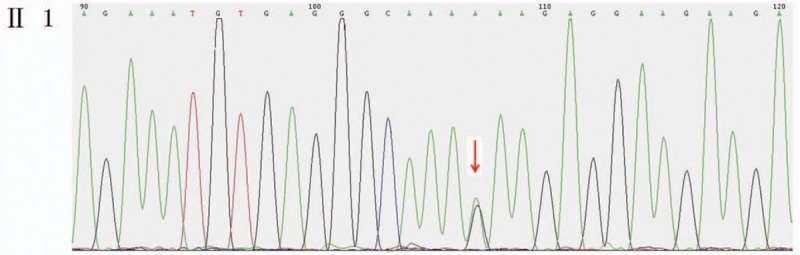
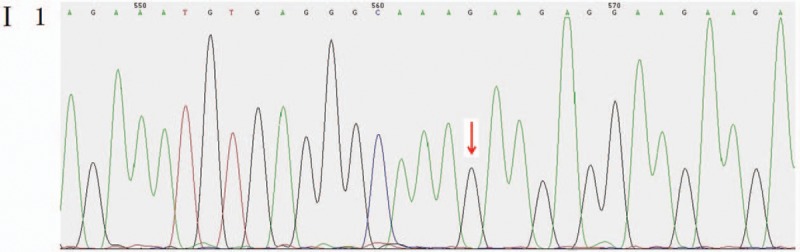
Outcomes: Gene detection revealed there to be a heterozygous mutation in the CHD7 gene of the patient, which was a missense mutation c.6571G > A (p.E2191K). The father's genotype was wild type, whereas it was the mutant type for the mother and younger brother. The distribution frequency of this mutation was zero in the dbSNP database, Hapmap, 1000 genomes database, and ExAC. Neither the mother nor the younger brother showed any clinical feature of KS or CHARGE syndrome.
Lessons: This study reports 1 case of KS with some clinical features of CHARGE syndrome as determined via clinical and genetic analysis, and found a new mutation in the CHD7 gene, suggesting that KS has an incomplete penetrance. Meanwhile, data suggested that mutation in the CHD7 gene could be detected in the setting of incomplete clinical manifestations of CHARGE syndrome, or without the usually believed manifestations of combined deafness as well as morphological abnormalities of the ear, providing new evidence for the differential diagnosis of KS with CHARGE syndrome in the future.
Conflict of interest statement
The authors report no conflicts of interest.
Figures
References
-
- Topaloglu AK, Kotan LD. Molecular causes of hypogonadotropic hypogonadism. Curr Opin Obstet Gynecol 2010;22:264–70. - PubMed
-
- Rugarli EL, Ballabio A. Kallmann syndrome: from genetics to neuro-biology. JAMA 1993;270:2713–6. - PubMed
-
- Kallmann FJ, Schoenfeld WA, Barrera SE. The genetic aspects of primary eunuchoidism. Am J Ment Defic 1944;48:203–36.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
