

A novel mutation causing type 1 Gaucher disease found in a Japanese patient with gastric cancer: A case report
- PMID: 29979419
- PMCID: PMC6076040
- DOI: 10.1097/MD.0000000000011361
A novel mutation causing type 1 Gaucher disease found in a Japanese patient with gastric cancer: A case report
Abstract
Rationale: Gaucher disease (GD) is an autosomal recessive disorder that leads to multiorgan complications caused by β-glucocerebrosidase deficiency due to mutations in the β-glucocerebrosidase-encoding gene (GBA). GD morbidity in Japan is quite rare and clinical phenotype and gene mutation patterns of patients with GD in Japan and Western countries differ considerably. Of Japanese patients with GD, 57% develop types 2 or 3 GD with neurologic manifestations and younger onset, whereas only 6% of patients with GD develop those manifestations in Western countries. Thus, it is relatively difficult to find and diagnose GD in Japan.
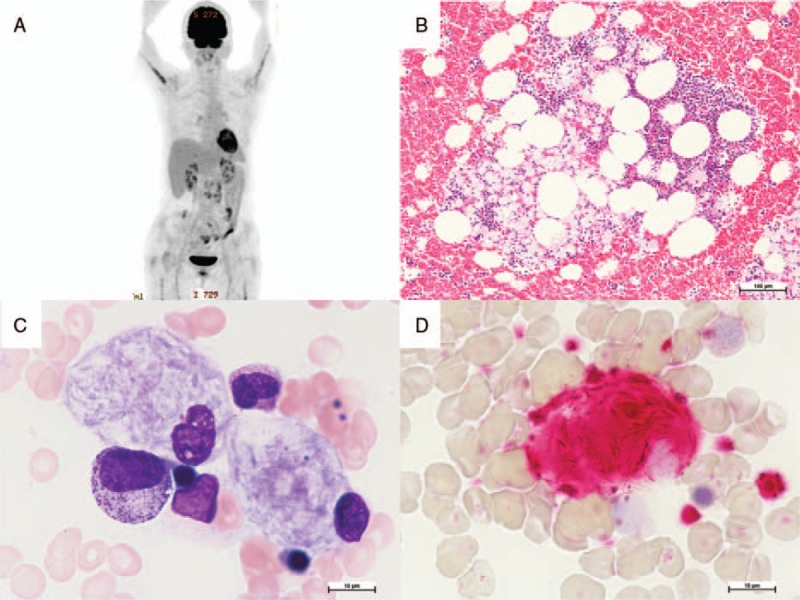
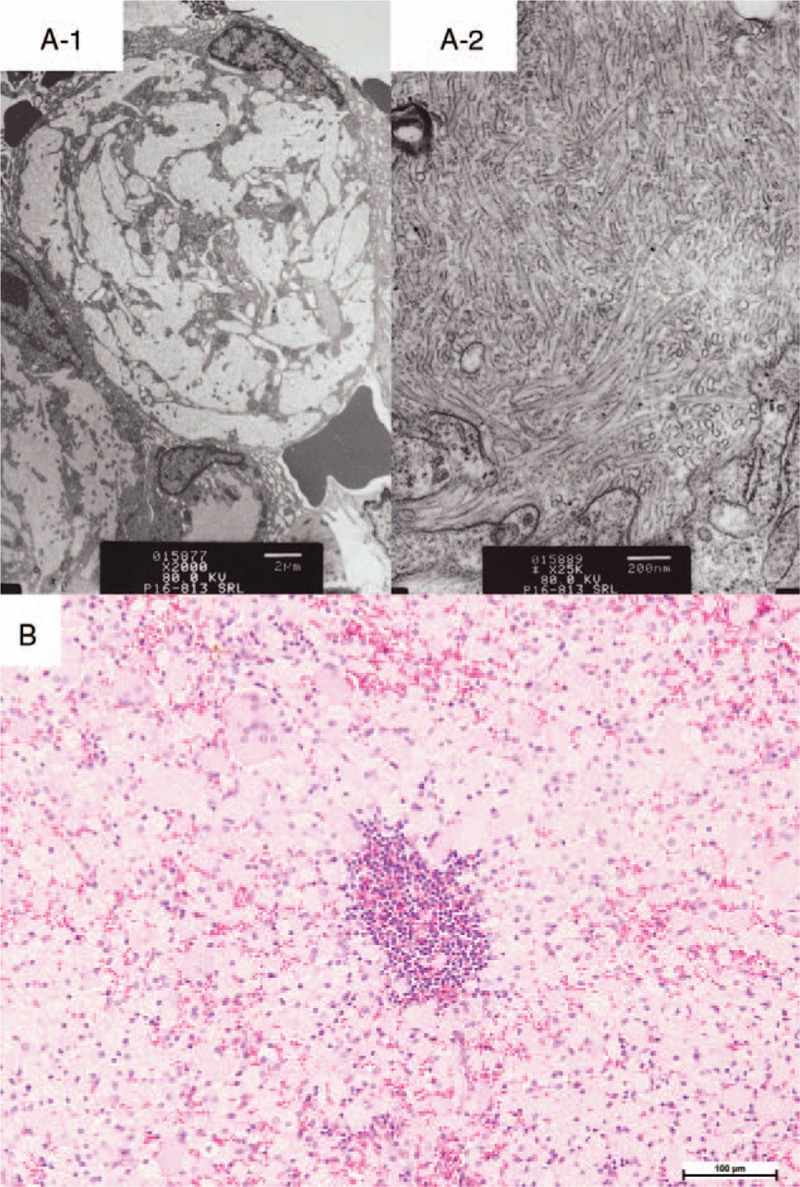
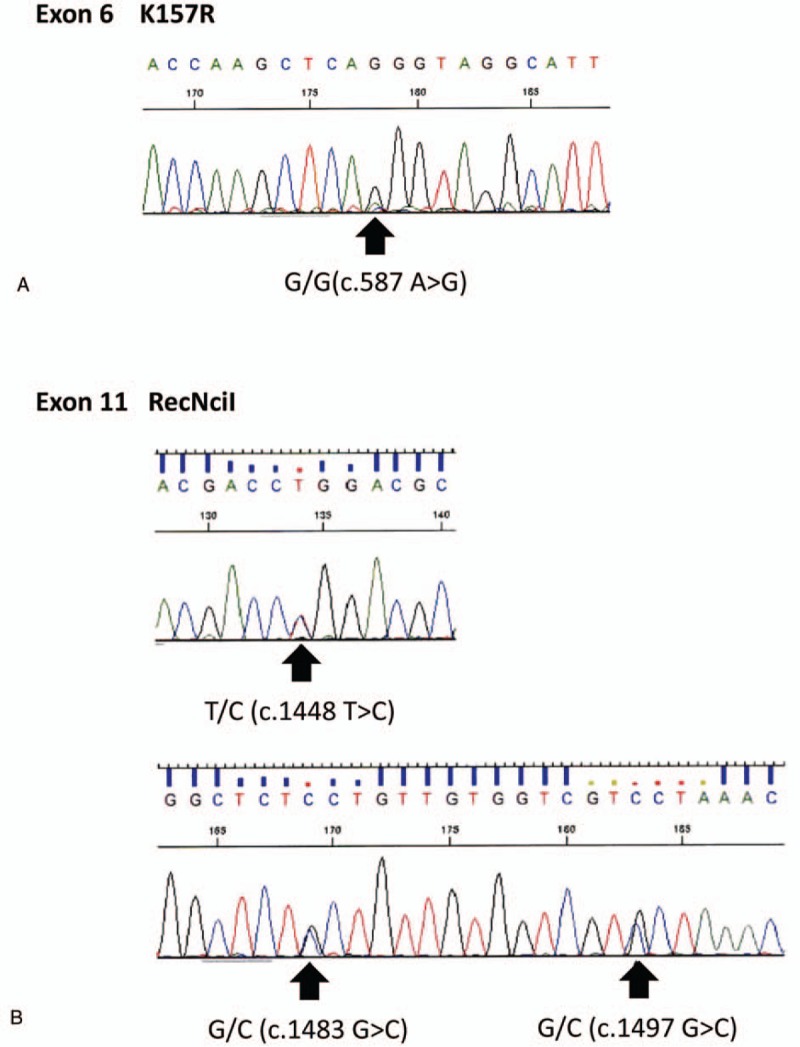
Patient concerns: A 69-year-old Japanese female with mild anemia and thrombocytopenia but without neurologic symptoms was initially referred for gastric cancer. Preoperative F-deoxyglucose positron emission tomography/computed tomography (FDG PET/CT) showed accumulation in the bone marrow and paraabdominal lymph nodes. Following bone marrow aspiration found, abnormal foamy macrophages in the bone marrow and electron microscopy revealed that the macrophages were filled with tubular-form structures. Adding to these signs suggestive of a lysosomal disease, serum β-glucocerebrosidase activity test found decreased. Sequencing of the patient's GBA gene revealed a RecNciI recombinant mutation and the novel mutation K157R (c.587A>G).
Diagnoses: On the basis of these findings and clinical manifestations, the final diagnosis of type 1 GD was made.
Interventions: Enzyme replacement therapy (ERT) with velaglucerase α was started after the diagnosis of type 1 GD.
Outcomes: The patient's β-glucocerebrosidase activity as well as hemoglobin and platelet levels were restored by ERT without any side effects. Bone marrow aspirations 10 months after the start of the treatment with velaglucerase α showed reduction of Gaucher cells in bone marrow to 2% from 4% of total cellularity.
Lessons: This is the first report of F-FDG PET/CT application providing a clue for GD diagnosis. A novel mutation in GBA is described, which implies a potential pool of patients with GD with this mutation in Japan.
Conflict of interest statement
The authors disclose no conflicts of interest.
Figures
Similar articles
-
Three mutations of adult type 1 Gaucher disease found in a Chinese patient: A case report.Medicine (Baltimore). 2018 Nov;97(47):e13161. doi: 10.1097/MD.0000000000013161. Medicine (Baltimore). 2018. PMID: 30461613 Free PMC article.
-
An observational study to investigate the relationship between plasma glucosylsphingosine (lyso-Gb1) concentration and treatment outcomes of patients with Gaucher disease in Japan.Orphanet J Rare Dis. 2022 Nov 3;17(1):401. doi: 10.1186/s13023-022-02549-6. Orphanet J Rare Dis. 2022. PMID: 36329499 Free PMC article.
-
Neuronopathic Gaucher disease presenting with microcytic hypochromic anemia.Int J Hematol. 2019 Mar;109(3):361-365. doi: 10.1007/s12185-018-2559-3. Epub 2018 Nov 19. Int J Hematol. 2019. PMID: 30456712
-
Simultaneous detection of Gaucher's disease and renal involvement of non-Hodgkin's lymphoma: the first Asian case report and a review of literature.Ann Clin Lab Sci. 2012 Summer;42(3):293-301. Ann Clin Lab Sci. 2012. PMID: 22964618 Review.
-
Velaglucerase alfa, a human recombinant glucocerebrosidase enzyme replacement therapy for type 1 Gaucher disease.Curr Opin Investig Drugs. 2010 Apr;11(4):472-8. Curr Opin Investig Drugs. 2010. PMID: 20336596 Review.
Cited by
-
Hematological Findings in Lysosomal Storage Disorders: A Perspective from the Medical Laboratory.EJIFCC. 2022 Apr 11;33(1):28-42. eCollection 2022 Apr. EJIFCC. 2022. PMID: 35645695 Free PMC article.
-
Diagnosis of Inherited Metabolic Disease in Older Patients: A Systematic Literature Review.J Inherit Metab Dis. 2025 May;48(3):e70038. doi: 10.1002/jimd.70038. J Inherit Metab Dis. 2025. PMID: 40406818 Free PMC article. Review.
-
Long-term safety and effectiveness of velaglucerase alfa in Gaucher disease: 6-year interim analysis of a post-marketing surveillance in Japan.Orphanet J Rare Dis. 2021 Dec 4;16(1):502. doi: 10.1186/s13023-021-02119-2. Orphanet J Rare Dis. 2021. PMID: 34863216 Free PMC article.
-
Screening for Gaucher Disease Using Dried Blood Spot Tests: A Japanese Multicenter, Cross-sectional Survey.Intern Med. 2021;60(5):699-707. doi: 10.2169/internalmedicine.5064-20. Epub 2021 Mar 1. Intern Med. 2021. PMID: 33642560 Free PMC article.
-
Three mutations of adult type 1 Gaucher disease found in a Chinese patient: A case report.Medicine (Baltimore). 2018 Nov;97(47):e13161. doi: 10.1097/MD.0000000000013161. Medicine (Baltimore). 2018. PMID: 30461613 Free PMC article.
References
-
- Brady RO, Kanfer JN, Shapiro D. Metabolism of glucocerebrosides. II. Evidence of an enzymatic deficiency in Gaucher's disease. Biochem Biophys Res Commun 1965;18:221–5. - PubMed
-
- Cox TM, Schofield JP. Gaucher's disease: clinical features and natural history. Baillieres Clin Haematol 1997;10:657–89. - PubMed
-
- Thomas AS, Mehta A, Hughes DA. Gaucher disease: haematological presentations and complications. Br J Haematol 2014;165:427–40. - PubMed
-
- Charrow J, Esplin JA, Gribble TJ, et al. Gaucher disease: recommendations on diagnosis, evaluation, and monitoring. Arch Intern Med 1998;158:1754–60. - PubMed
-
- Luan Z, Li L, Higaki K, et al. The chaperone activity and toxicity of ambroxol on Gaucher cells and normal mice. Brain Dev 2013;35:317–22. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
