

Mechanism-based inhibition of human persulfide dioxygenase by γ-glutamyl-homocysteinyl-glycine
- PMID: 29980601
- PMCID: PMC6093238
- DOI: 10.1074/jbc.RA118.004096
Mechanism-based inhibition of human persulfide dioxygenase by γ-glutamyl-homocysteinyl-glycine
Abstract
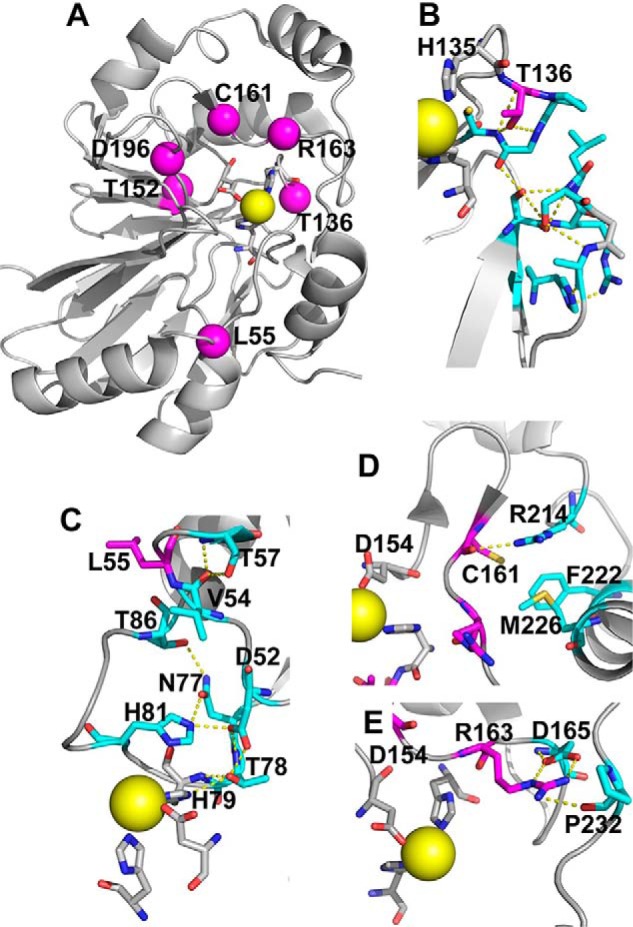
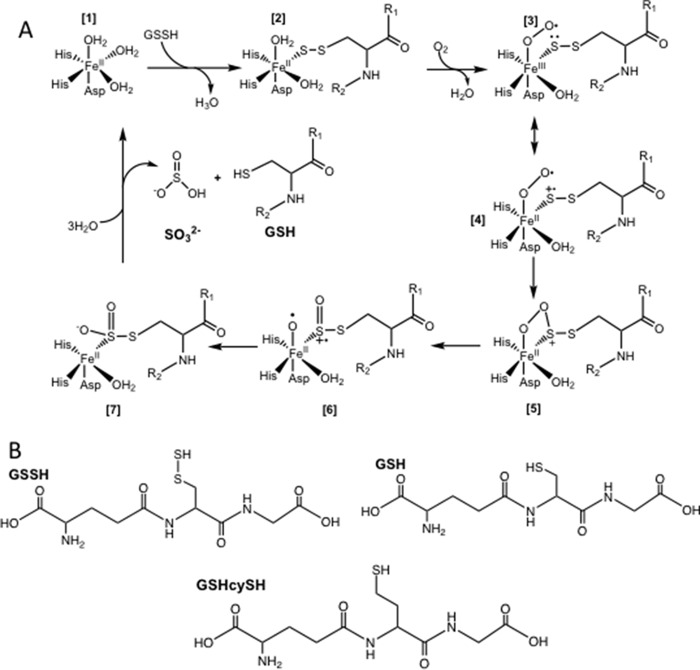
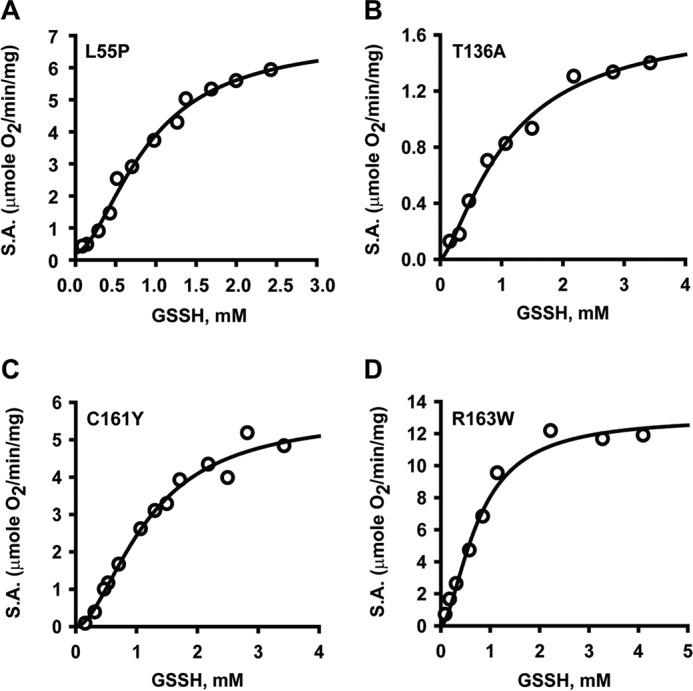
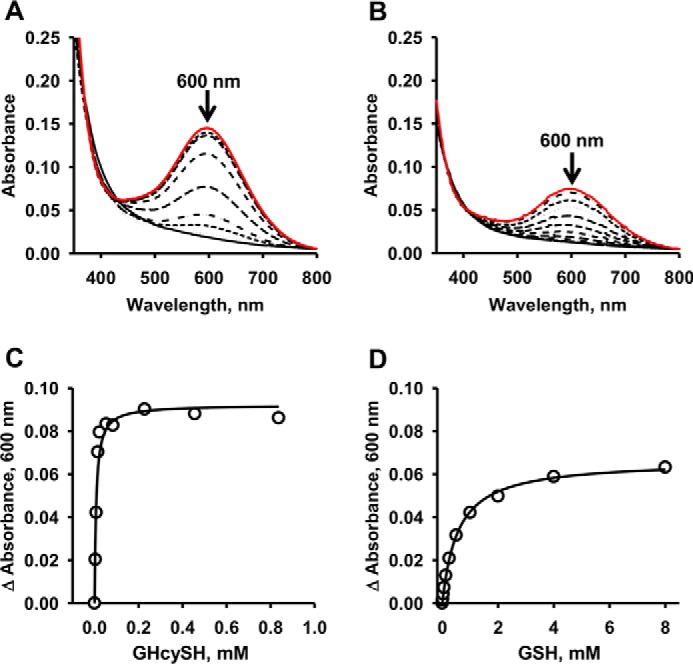
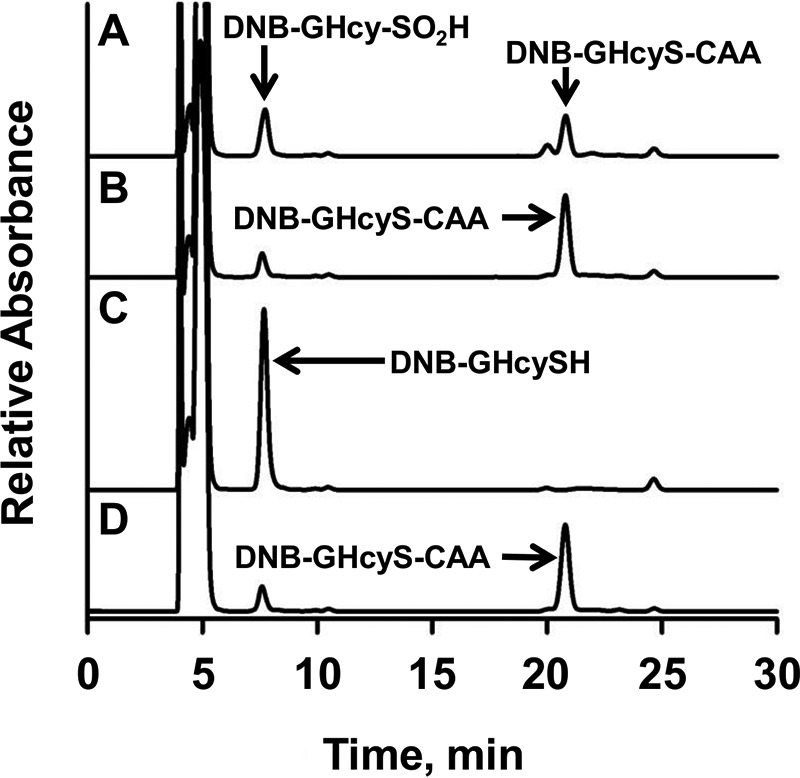
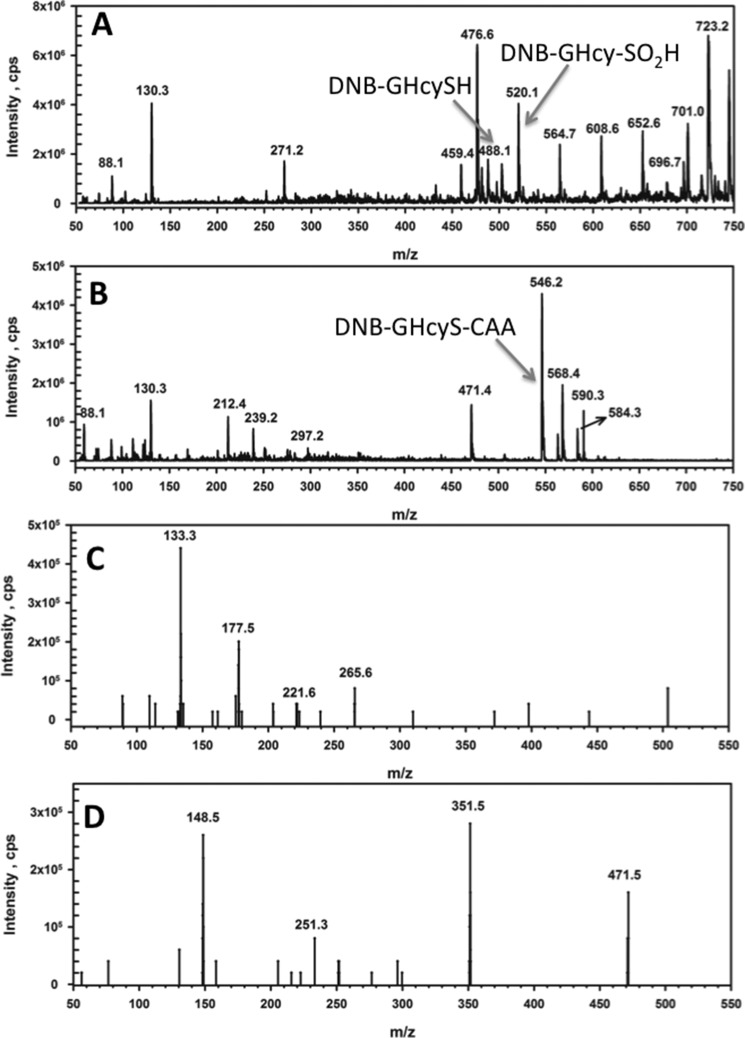
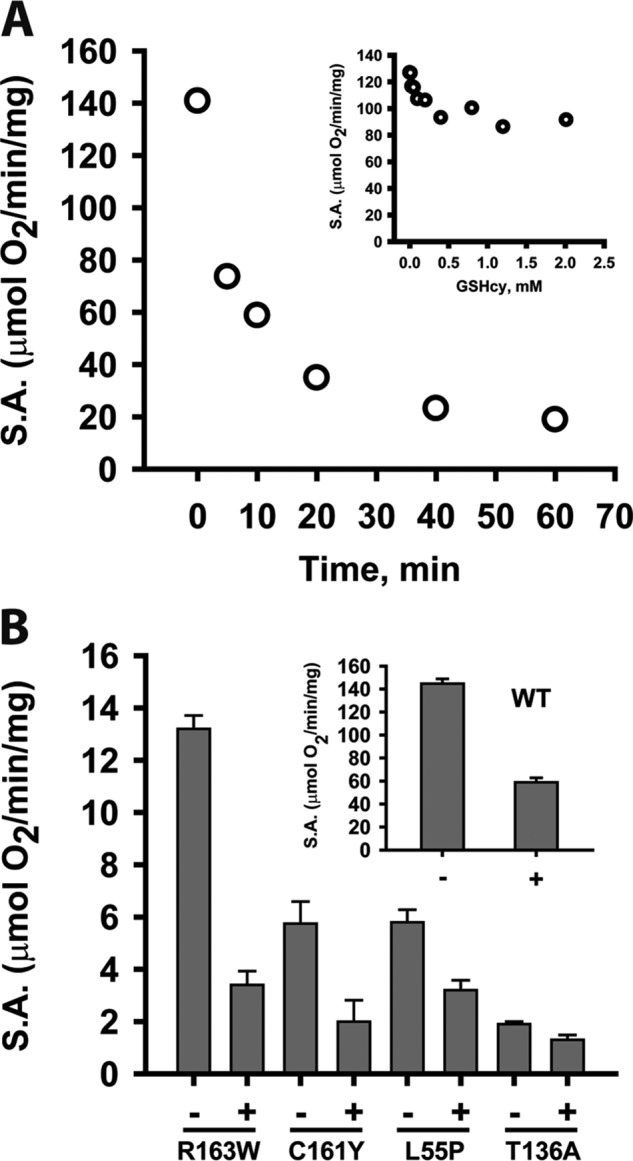
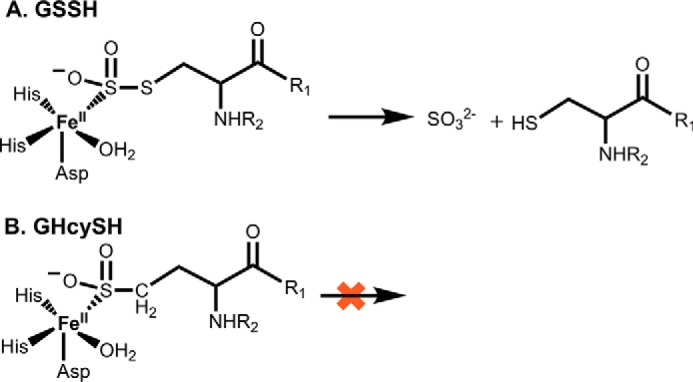
Hydrogen sulfide (H2S) is a signaling molecule with many beneficial effects. However, its cellular concentration is strictly regulated to avoid toxicity. Persulfide dioxygenase (PDO or ETHE1) is a mononuclear non-heme iron-containing protein in the sulfide oxidation pathway catalyzing the conversion of GSH persulfide (GSSH) to sulfite and GSH. PDO mutations result in the autosomal-recessive disorder ethylmalonic encephalopathy (EE). Here, we developed γ-glutamyl-homocysteinyl-glycine (GHcySH), in which the cysteinyl moiety in GSH is substituted with homocysteine, as a mechanism-based PDO inhibitor. Human PDO used GHcySH as an alternative substrate and converted it to GHcy-SO2H, mimicking GS-SO2H, the putative oxygenated intermediate formed with the natural substrate. Because GHcy-SO2H contains a C-S bond rather than an S-S bond in GS-SO2H, it failed to undergo the final hydrolysis step in the catalytic cycle, leading to PDO inhibition. We also characterized the biochemical penalties incurred by the L55P, T136A, C161Y, and R163W mutations reported in EE patients. The variants displayed lower iron content (1.4-11-fold) and lower thermal stability (1.2-1.7-fold) than WT PDO. They also exhibited varying degrees of catalytic impairment; the kcat/Km values for R163W, L55P, and C161Y PDOs were 18-, 42-, and 65-fold lower, respectively, and the T136A variant was most affected, with a 200-fold lower kcat/Km Like WT enzyme, these variants were inhibited by GHcySH. This study provides the first characterization of an intermediate in the PDO-catalyzed reaction and reports on deficits associated with EE-linked mutations that are distal from the active site.
Keywords: dioxygenase; enzyme kinetics; enzyme mechanism; ethylmalonic acid; ethylmalonic encephalopathy; hydrogen sulfide; iron; persulfide dioxygenase; sulfide oxidation.
© 2018 Kabil et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- García-Silva M. T., Campos Y., Ribes A., Briones P., Cabello A., Santos Borbujo J., Arenas J., and Garavaglia B. (1994) Encephalopathy, petechiae, and acrocyanosis with ethylmalonic aciduria associated with muscle cytochrome c oxidase deficiency. J. Pediatr. 125, 843–844 10.1016/S0022-3476(06)80197-6 - DOI - PubMed
-
- Tiranti V., Briem E., Lamantea E., Mineri R., Papaleo E., De Gioia L., Forlani F., Rinaldo P., Dickson P., Abu-Libdeh B., Cindro-Heberle L., Owaidha M., Jack R. M., Christensen E., Burlina A., and Zeviani M. (2006) ETHE1 mutations are specific to ethylmalonic encephalopathy. J. Med. Genet. 43, 340–346 - PMC - PubMed
-
- Tiranti V., D'Adamo P., Briem E., Ferrari G., Mineri R., Lamantea E., Mandel H., Balestri P., Garcia-Silva M. T., Vollmer B., Rinaldo P., Hahn S. H., Leonard J., Rahman S., Dionisi-Vici C., et al. (2004) Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. Am. J. Hum. Genet. 74, 239–252 10.1086/381653 - DOI - PMC - PubMed
-
- Tiranti V., Viscomi C., Hildebrandt T., Di Meo I., Mineri R., Tiveron C., Levitt M. D., Prelle A., Fagiolari G., Rimoldi M., and Zeviani M. (2009) Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 15, 200–205 10.1038/nm.1907 - DOI - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
