

Intermedin protects against sepsis by concurrently re-establishing the endothelial barrier and alleviating inflammatory responses
- PMID: 29980671
- PMCID: PMC6035189
- DOI: 10.1038/s41467-018-05062-2
Intermedin protects against sepsis by concurrently re-establishing the endothelial barrier and alleviating inflammatory responses
Abstract
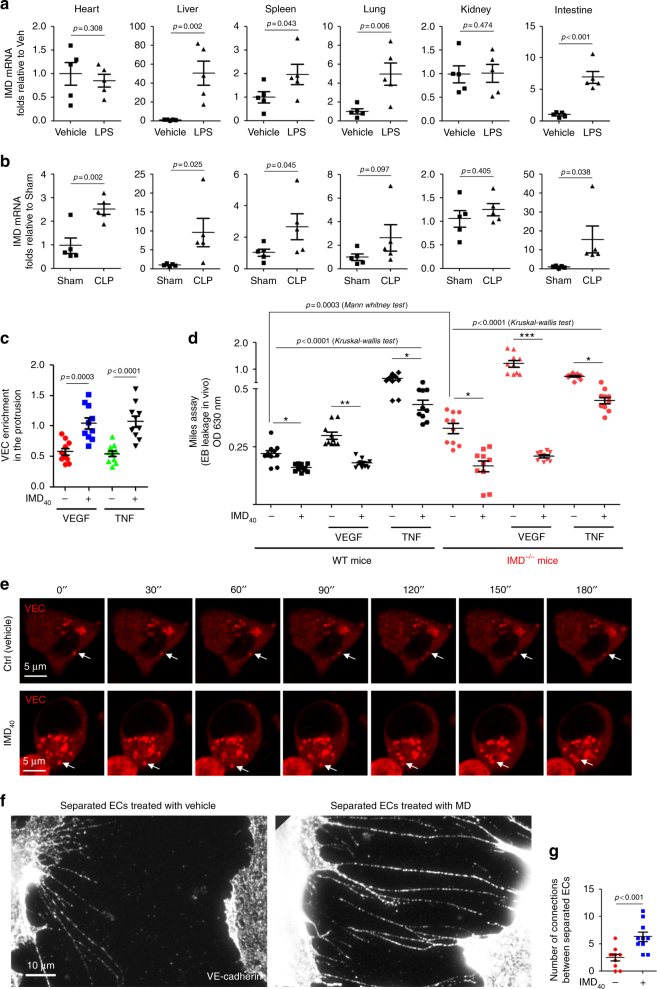
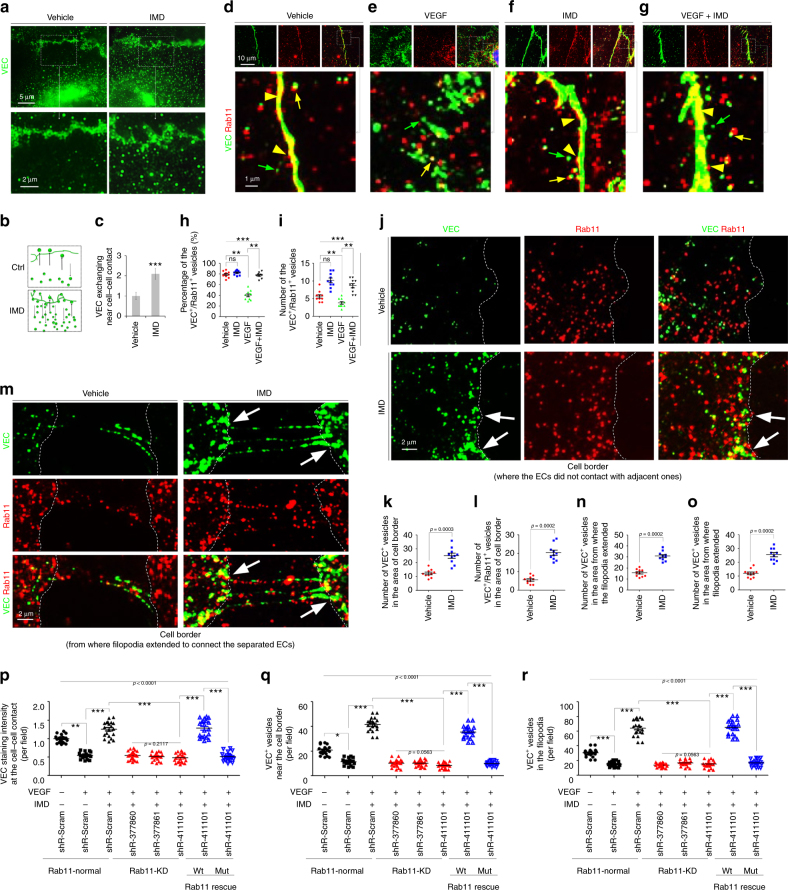
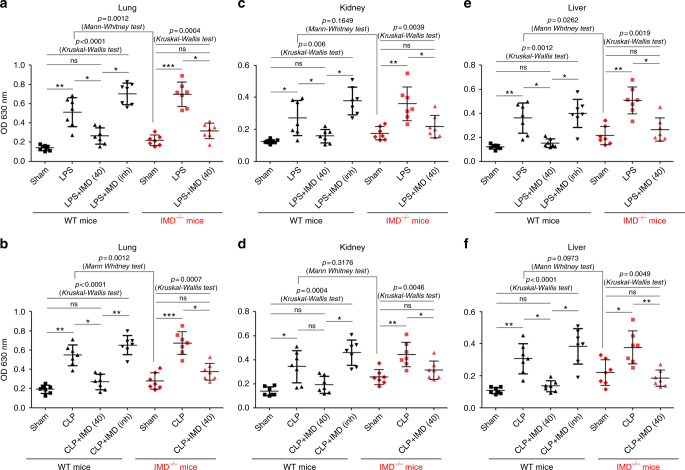
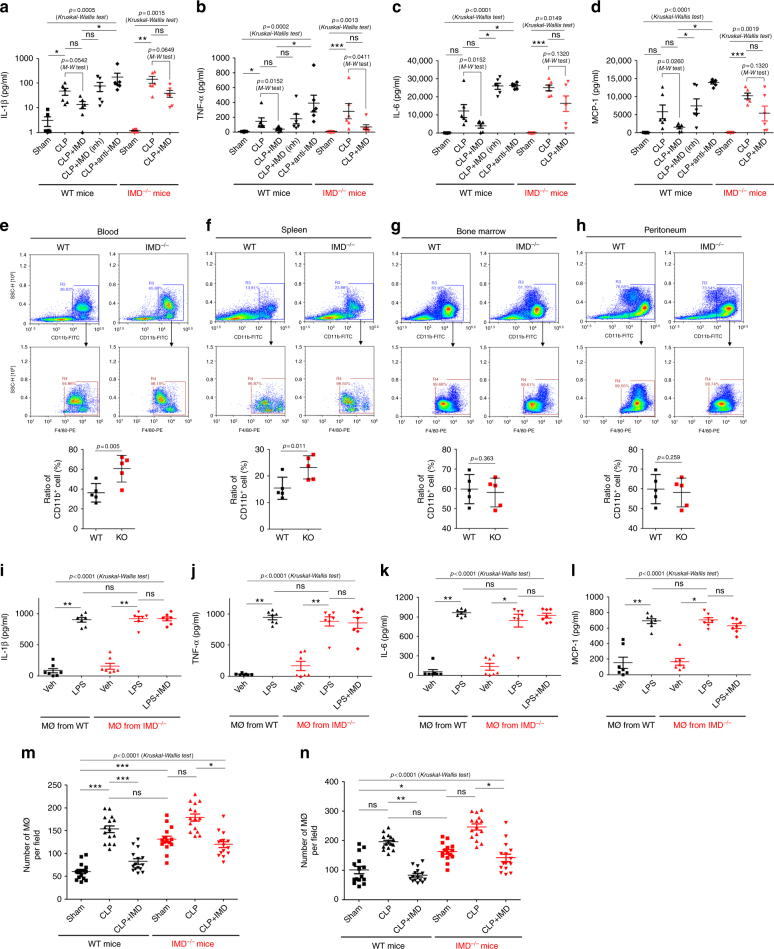
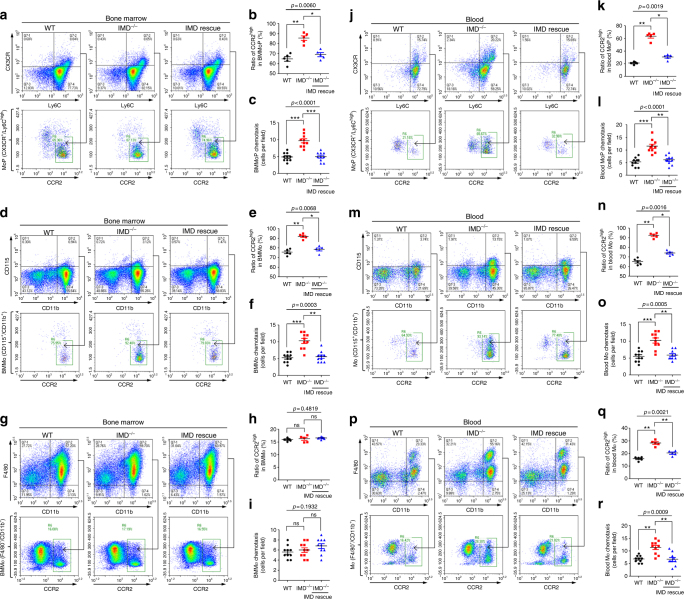
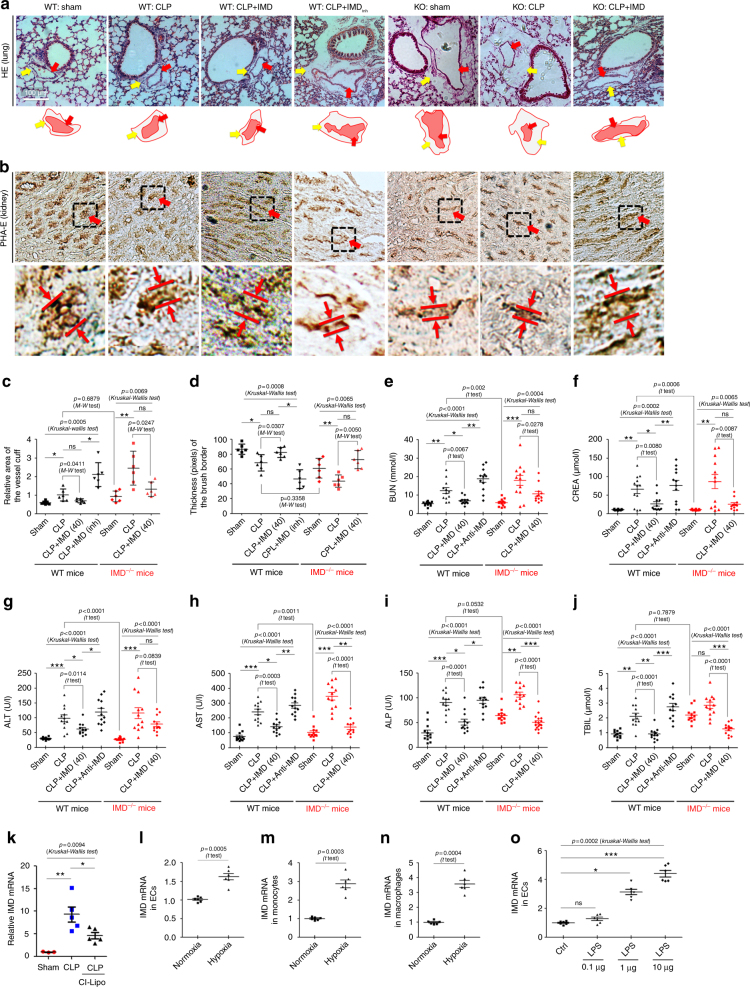
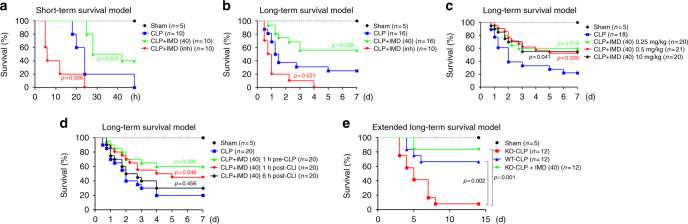
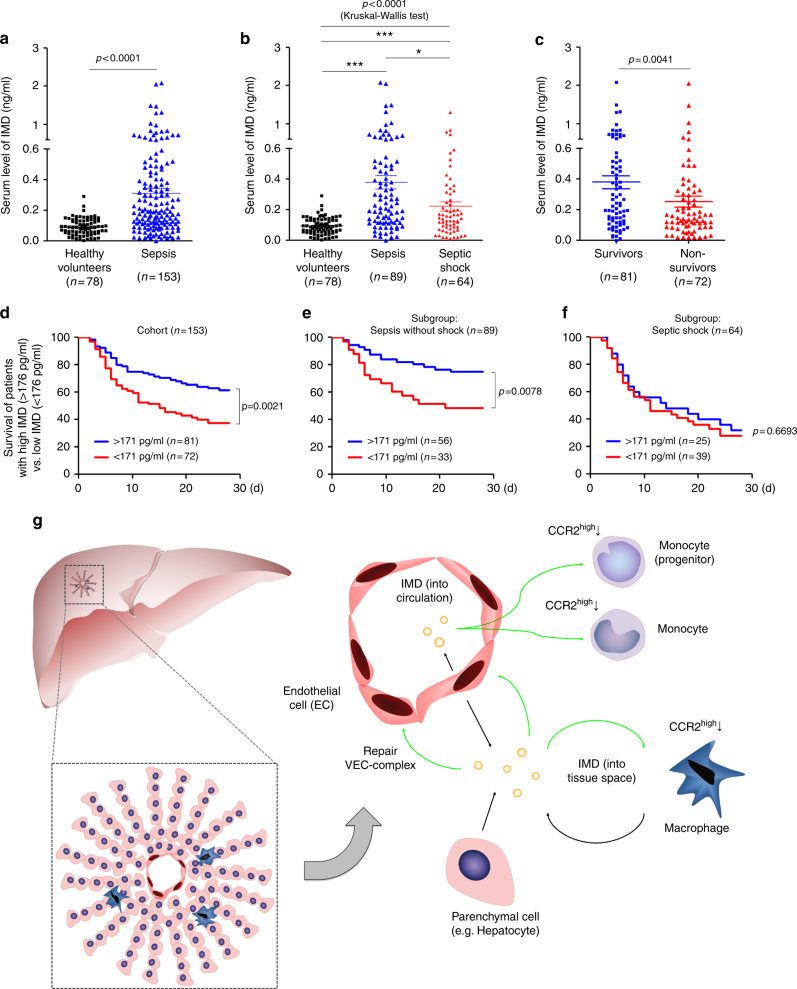
Sepsis is a life-threatening condition caused by dysregulated host responses to infection. Widespread vascular hyperpermeability and a "cytokine storm" are two pathophysiological hallmarks of sepsis. Here, we show that intermedin (IMD), a member of the calcitonin family, alleviates organ injury and decreases mortality in septic mice by concurrently alleviating vascular leakage and inflammatory responses. IMD promotes the relocation of vascular endothelial cadherin through a Rab11-dependent pathway to dynamically repair the disrupted endothelial junction. Additionally, IMD decreases inflammatory responses by reducing macrophage infiltration via downregulating CCR2 expression. IMD peptide administration ameliorates organ injuries and significantly improves the survival of septic mice, and the experimental results correlate with the clinical data. Patients with high IMD levels exhibit a lower risk of shock, lower severity scores, and greatly improved survival outcomes than those with low IMD levels. Based on our data, IMD may be an important self-protective factor in response to sepsis.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
