

Disentangling the drivers of functional complexity at the metagenomic level in Shark Bay microbial mat microbiomes
- PMID: 29980796
- PMCID: PMC6194083
- DOI: 10.1038/s41396-018-0208-8
Disentangling the drivers of functional complexity at the metagenomic level in Shark Bay microbial mat microbiomes
Abstract
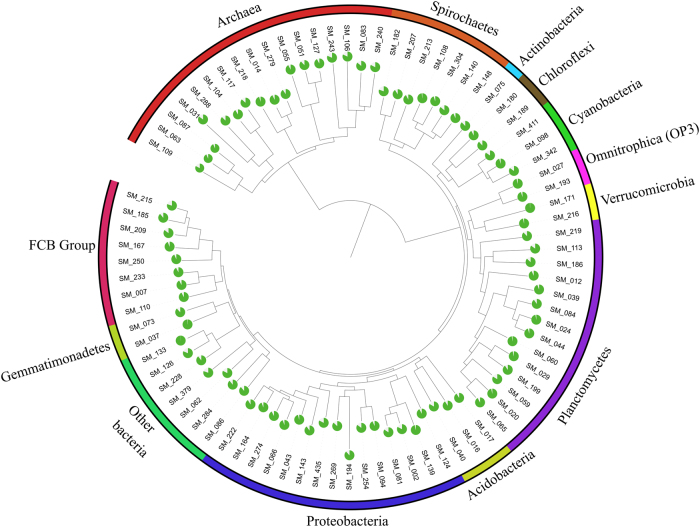
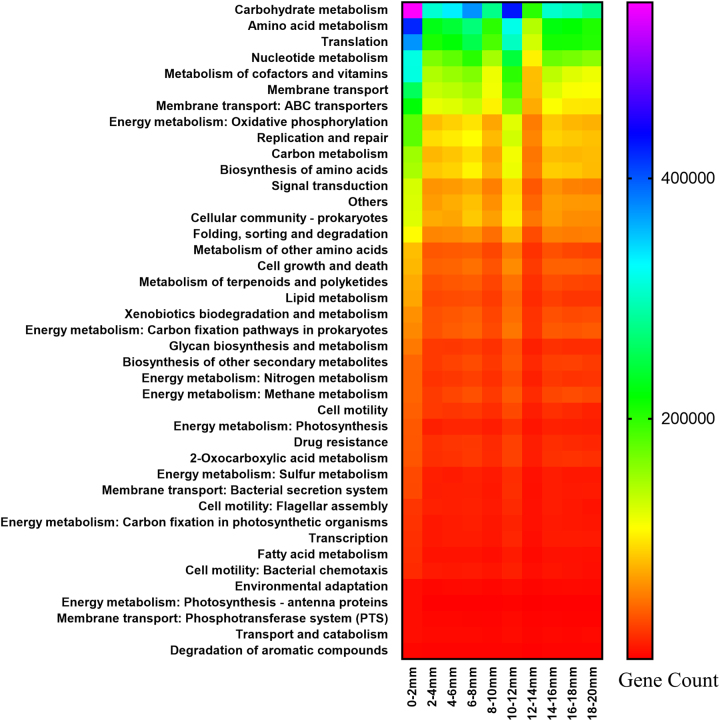
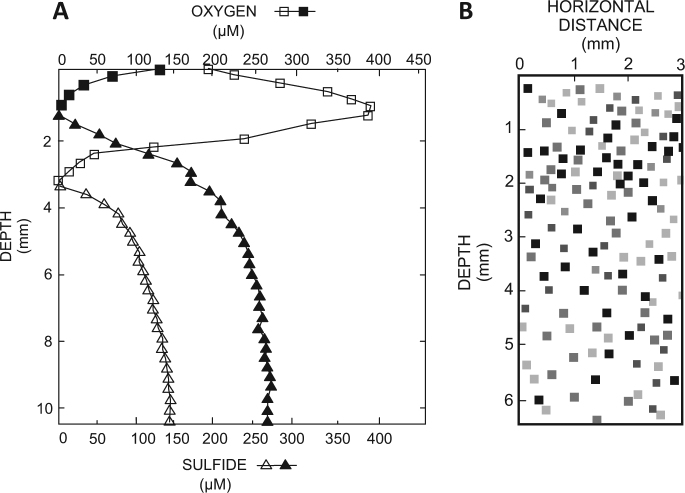
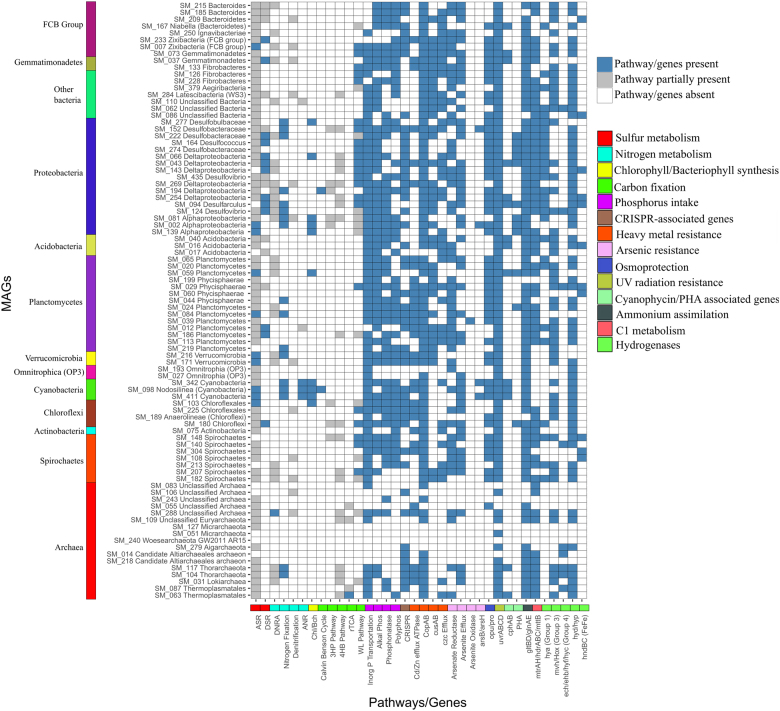
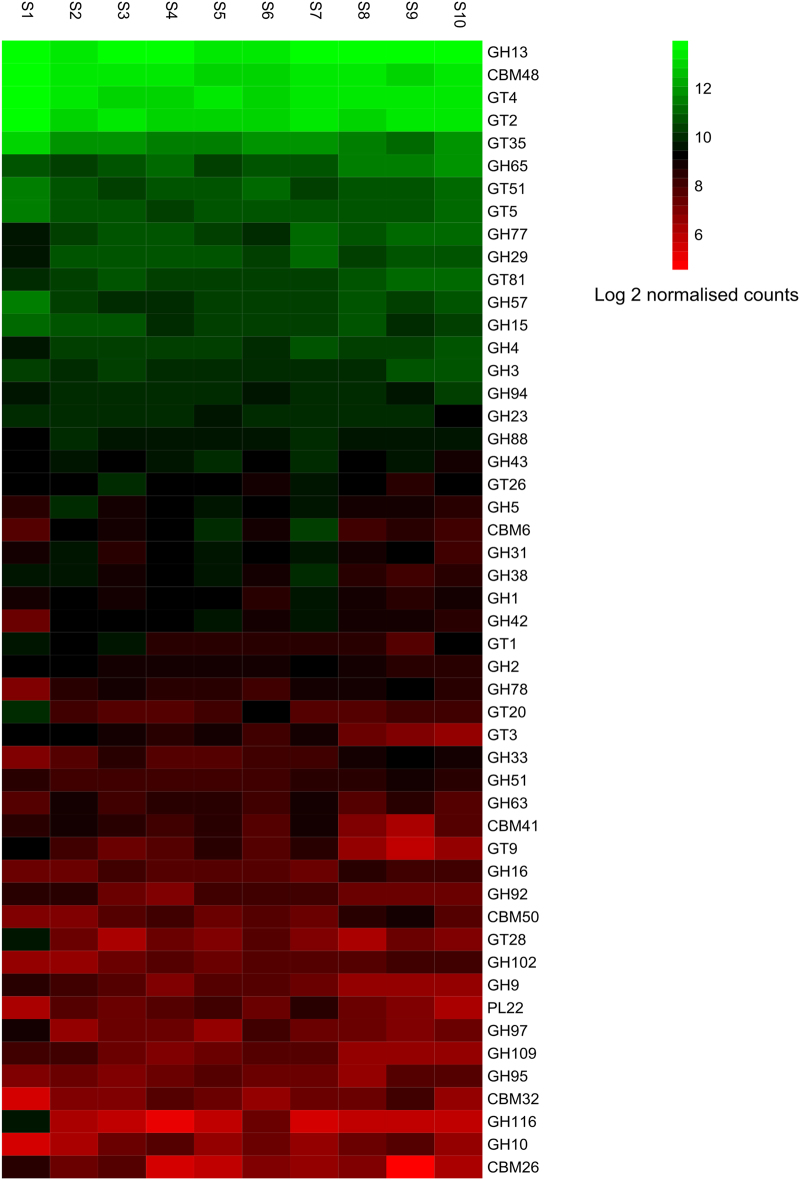
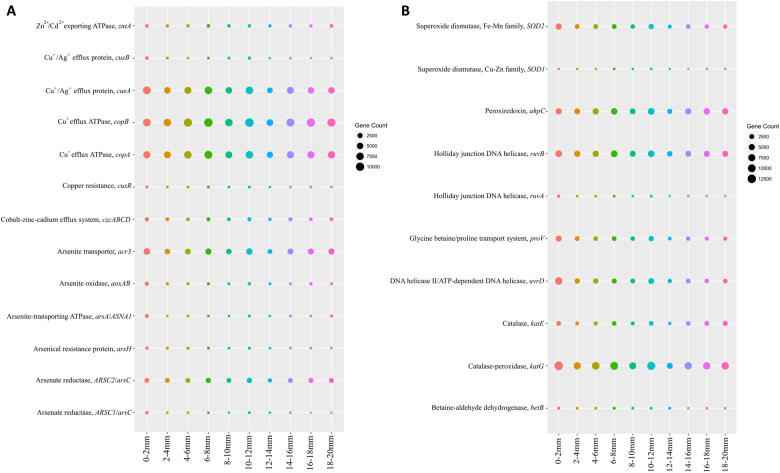
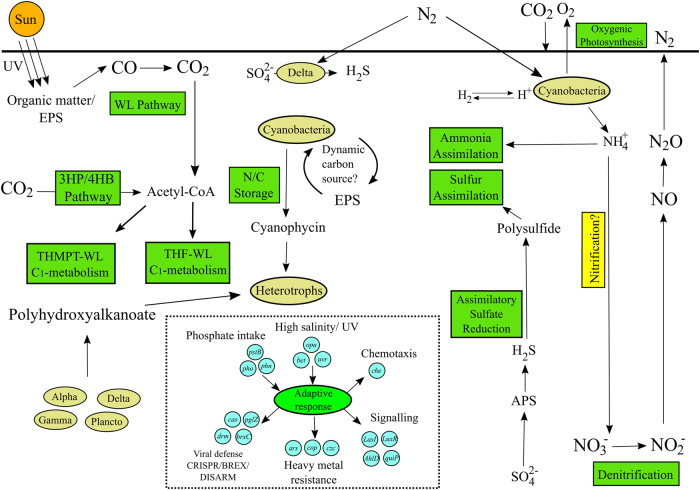
The functional metagenomic potential of Shark Bay microbial mats was examined for the first time at a millimeter scale, employing shotgun sequencing of communities via the Illumina NextSeq 500 platform in conjunction with defined chemical analyses. A detailed functional metagenomic profile has elucidated key pathways and facilitated inference of critical microbial interactions. In addition, 87 medium-to-high-quality metagenome-assembled genomes (MAG) were assembled, including potentially novel bins under the deep-branching archaeal Asgard group (Thorarchaetoa and Lokiarchaeota). A range of pathways involved in carbon, nitrogen, sulfur, and phosphorus cycles were identified in mat metagenomes, with the Wood-Ljungdahl pathway over-represented and inferred as a major carbon fixation mode. The top five sets of genes were affiliated to sulfate assimilation (cysNC cysNCD, sat), methanogenesis (hdrABC), Wood-Ljungdahl pathways (cooS, coxSML), phosphate transport (pstB), and copper efflux (copA). Polyhydroxyalkanoate (PHA) synthase genes were over-represented at the surface, with PHA serving as a potential storage of fixed carbon. Sulfur metabolism genes were highly represented, in particular complete sets of genes responsible for both assimilatory and dissimilatory sulfate reduction. Pathways of environmental adaptation (UV, hypersalinity, oxidative stress, and heavy metal resistance) were also delineated, as well as putative viral defensive mechanisms (core genes of the CRISPR, BREX, and DISARM systems). This study provides new metagenome-based models of how biogeochemical cycles and adaptive responses may be partitioned in the microbial mats of Shark Bay.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

References
-
- Dupraz C, Visscher PT. Microbial lithification in marine stromatolites and hypersaline mats. Trends Microbiol. 2005;13:429–38. - PubMed
-
- Allen MA, Goh F, Burns BP, Neilan BA. Bacterial, archaeal and eukaryotic diversity of smooth and pustular microbial mat communities in the hypersaline lagoon of Shark Bay. Geobiology. 2009;7:82–96. - PubMed
-
- Burns BP, Goh F, Allen M, Neilan BA. Microbial diversity of extant stromatolites in the hypersaline marine environment of Shark Bay, Australia. Environ Microbiol. 2004;6:1096. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
