

Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry
- PMID: 29988108
- PMCID: PMC6211289
- DOI: 10.1038/s41596-018-0006-9
Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry
Abstract
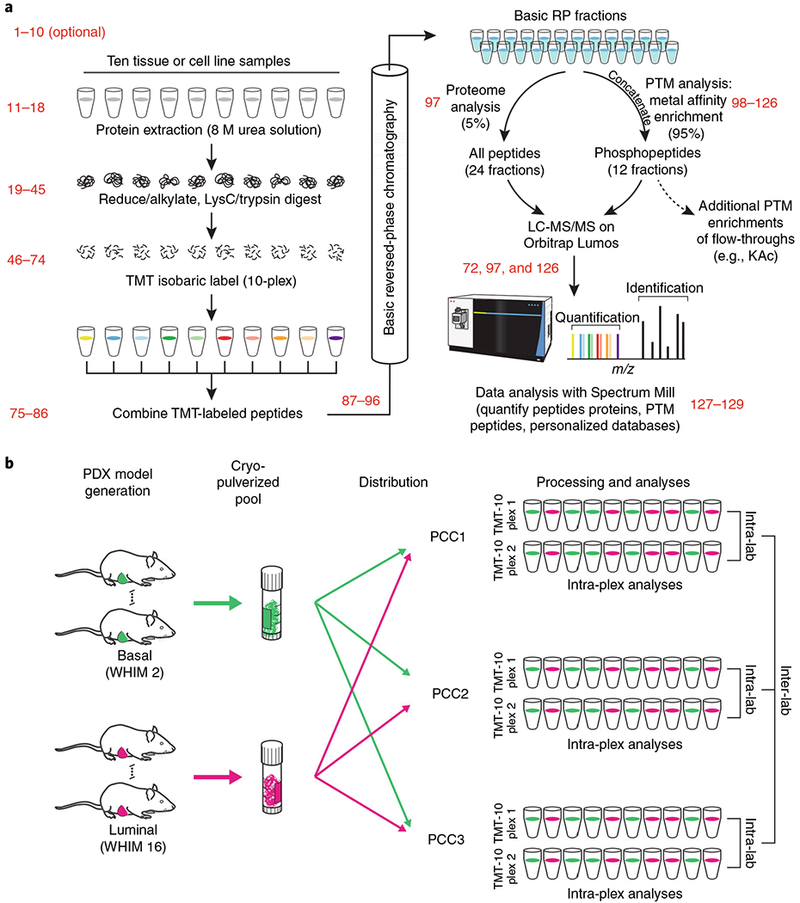
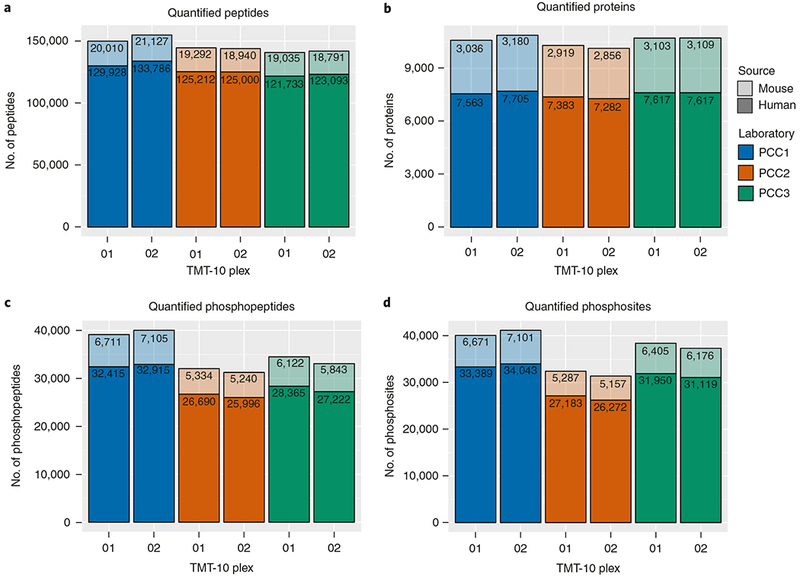
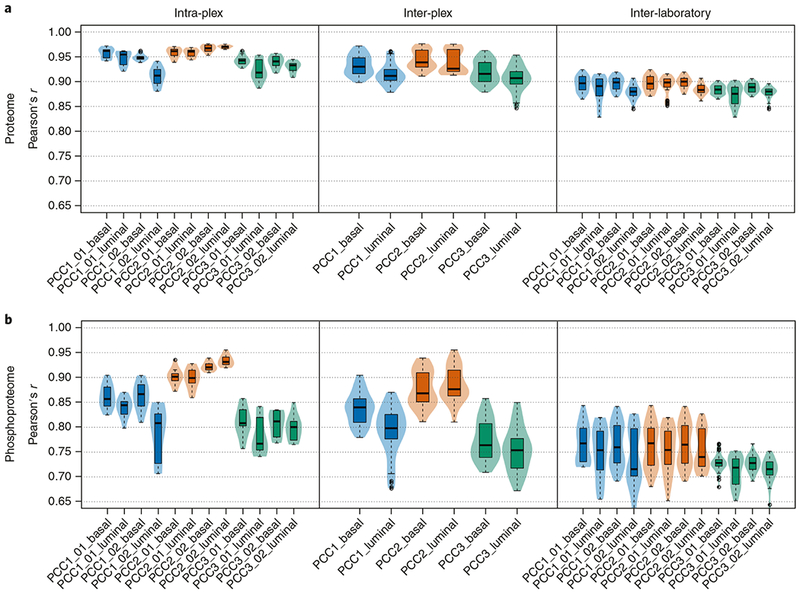
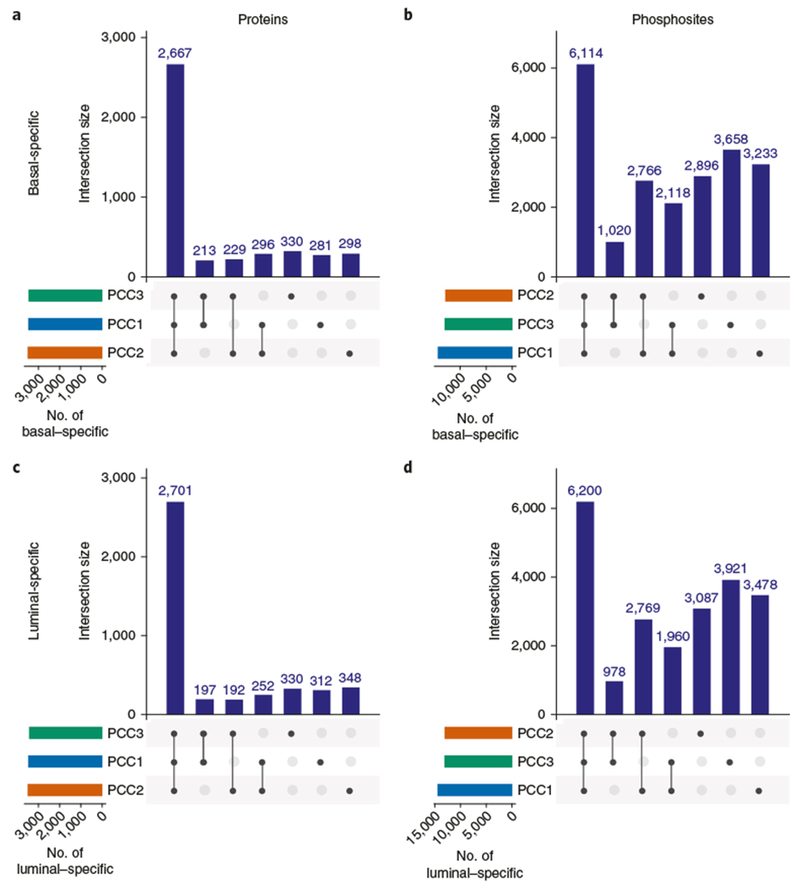
Here we present an optimized workflow for global proteome and phosphoproteome analysis of tissues or cell lines that uses isobaric tags (TMT (tandem mass tags)-10) for multiplexed analysis and relative quantification, and provides 3× higher throughput than iTRAQ (isobaric tags for absolute and relative quantification)-4-based methods with high intra- and inter-laboratory reproducibility. The workflow was systematically characterized and benchmarked across three independent laboratories using two distinct breast cancer subtypes from patient-derived xenograft models to enable assessment of proteome and phosphoproteome depth and quantitative reproducibility. Each plex consisted of ten samples, each being 300 μg of peptide derived from <50 mg of wet-weight tissue. Of the 10,000 proteins quantified per sample, we could distinguish 7,700 human proteins derived from tumor cells and 3100 mouse proteins derived from the surrounding stroma and blood. The maximum deviation across replicates and laboratories was <7%, and the inter-laboratory correlation for TMT ratio-based comparison of the two breast cancer subtypes was r > 0.88. The maximum deviation for the phosphoproteome coverage was <24% across laboratories, with an average of >37,000 quantified phosphosites per sample and differential quantification correlations of r > 0.72. The full procedure, including sample processing and data generation, can be completed within 10 d for ten tissue samples, and 100 samples can be analyzed in ~4 months using a single LC-MS/MS instrument. The high quality, depth, and reproducibility of the data obtained both within and across laboratories should enable new biological insights to be obtained from mass spectrometry-based proteomics analyses of cells and tissues together with proteogenomic data integration.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
