

Bi-allelic Loss of CDKN2A Initiates Melanoma Invasion via BRN2 Activation
- PMID: 29990501
- PMCID: PMC6084788
- DOI: 10.1016/j.ccell.2018.05.014
Bi-allelic Loss of CDKN2A Initiates Melanoma Invasion via BRN2 Activation
Abstract
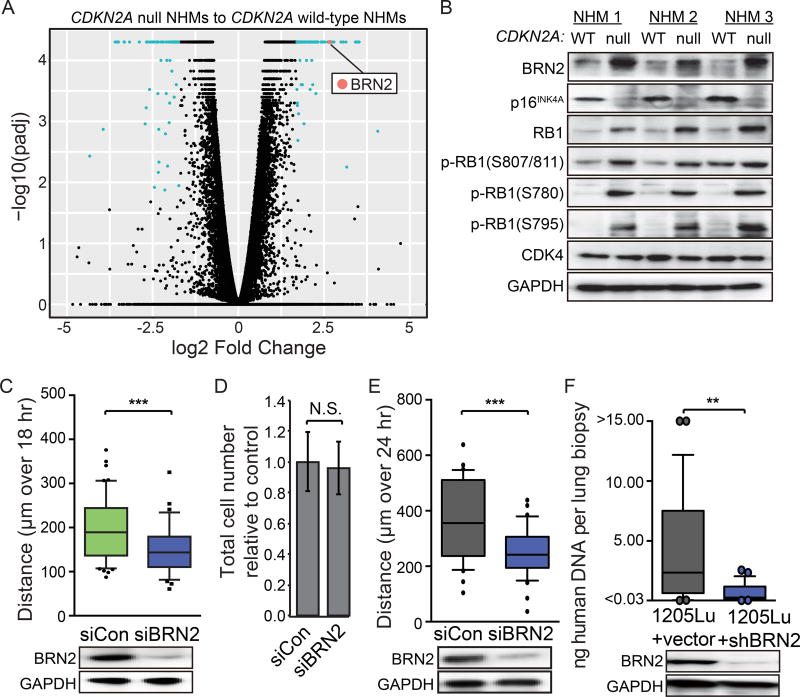
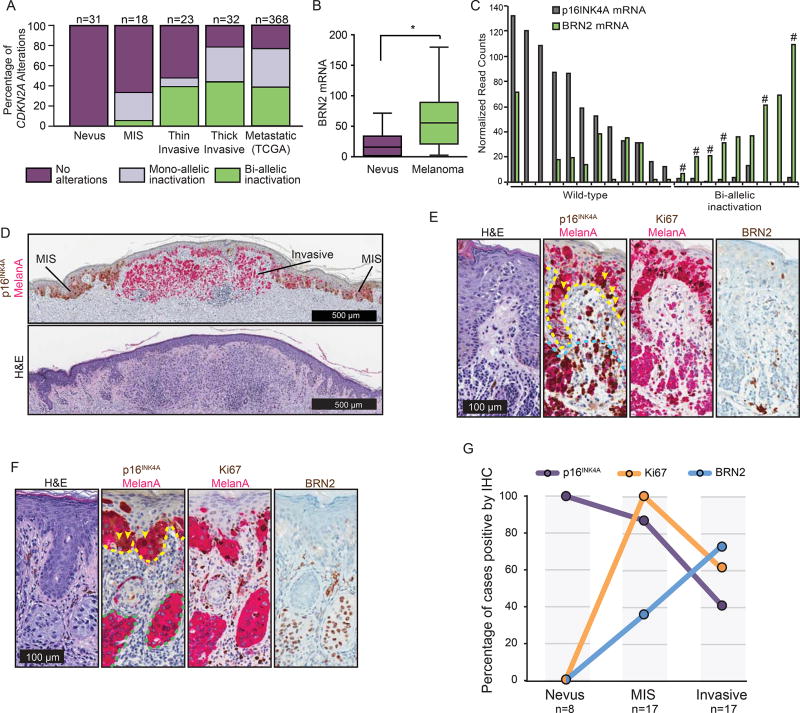
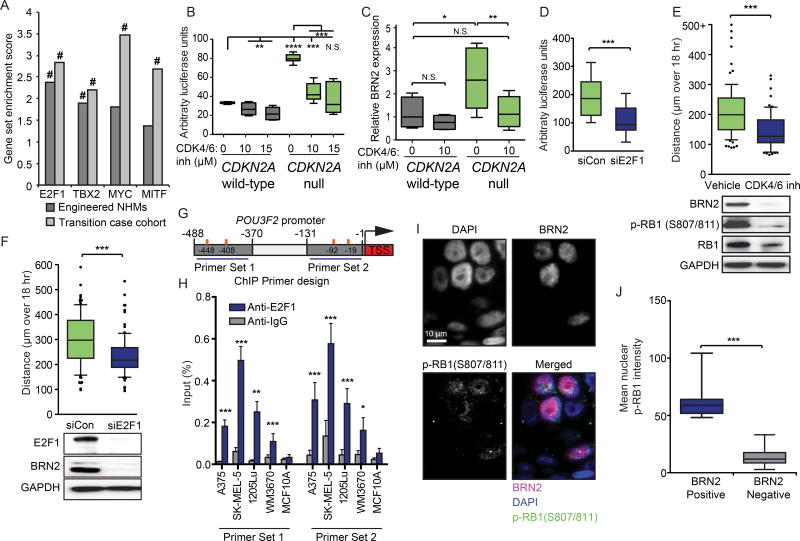
Loss of the CDKN2A tumor suppressor is associated with melanoma metastasis, but the mechanisms connecting the phenomena are unknown. Using CRISPR-Cas9 to engineer a cellular model of melanoma initiation from primary human melanocytes, we discovered that a lineage-restricted transcription factor, BRN2, is downstream of CDKN2A and directly regulated by E2F1. In a cohort of melanocytic tumors that capture distinct progression stages, we observed that CDKN2A loss coincides with both the onset of invasive behavior and increased BRN2 expression. Loss of the CDKN2A protein product p16INK4A permitted metastatic dissemination of human melanoma lines in mice, a phenotype rescued by inhibition of BRN2. These results demonstrate a mechanism by which CDKN2A suppresses the initiation of melanoma invasion through inhibition of BRN2.
Keywords: BRN2; CDKN2A; CRISPR engineering; E2F1; invasion; melanocytes; melanoma.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
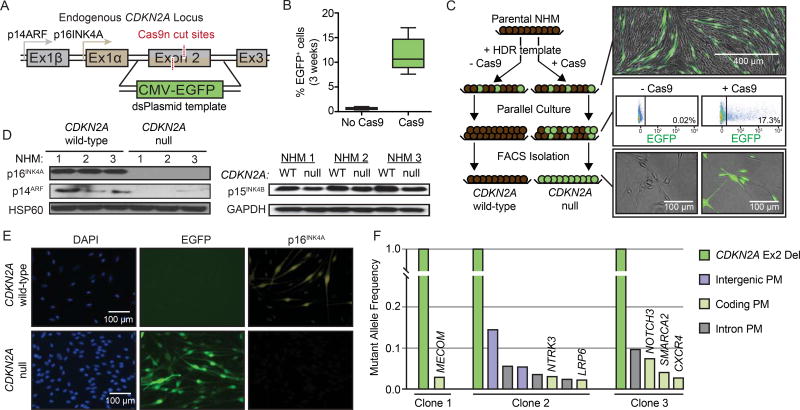
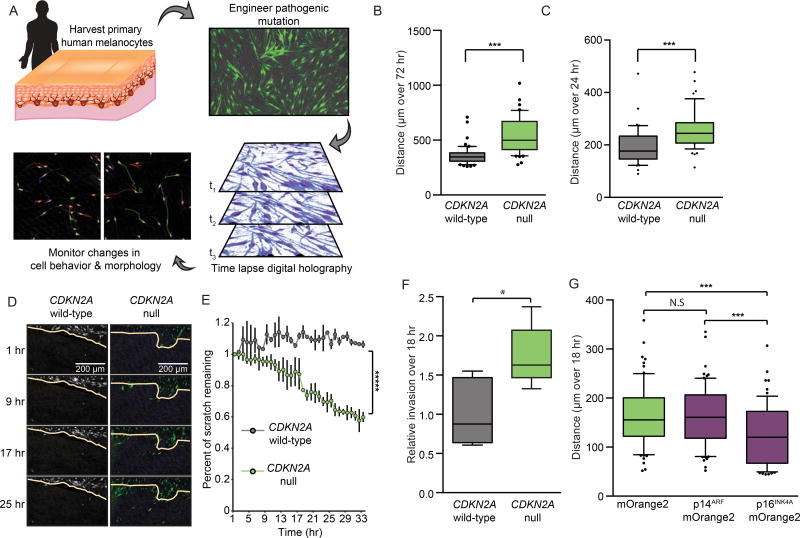
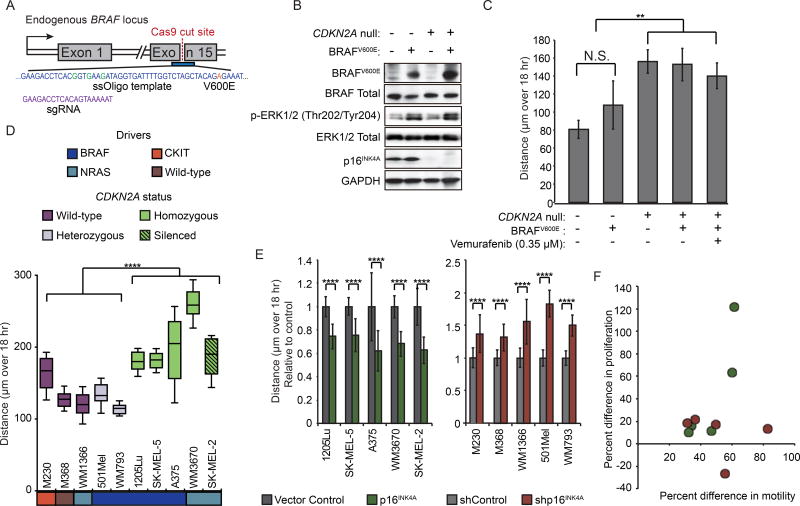
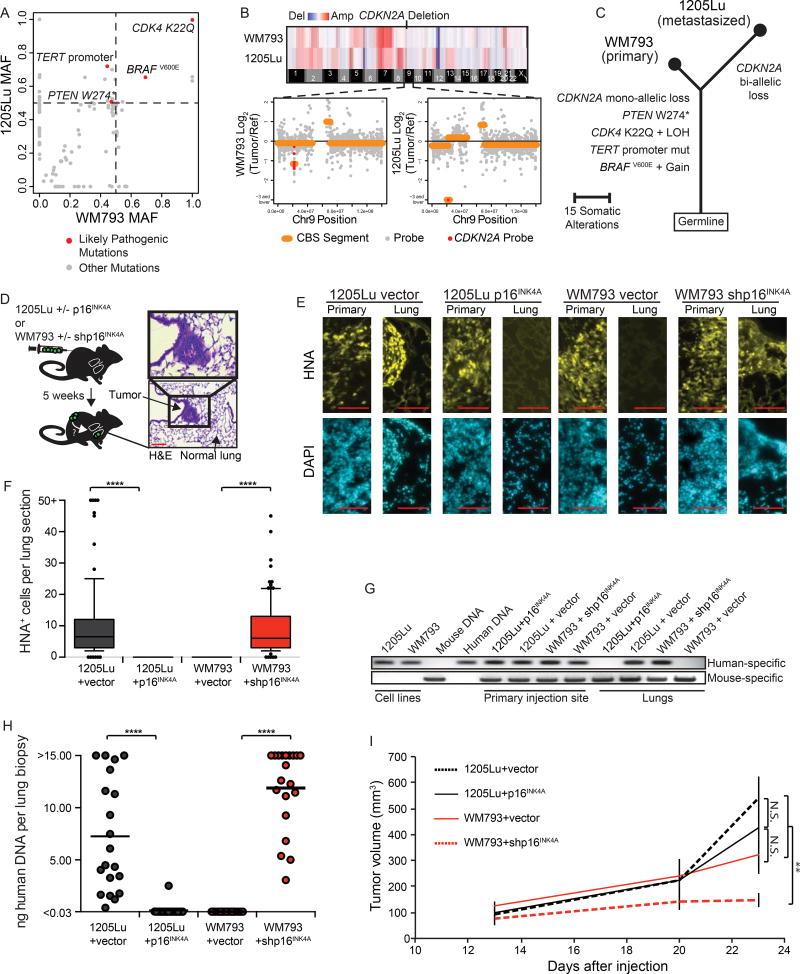
Comment in
-
BRN 2 Invade.Cancer Cell. 2018 Jul 9;34(1):1-3. doi: 10.1016/j.ccell.2018.06.010. Cancer Cell. 2018. PMID: 29990495 Free PMC article.
References
-
- Ackermann J, Frutschi M, Kaloulis K, McKee T, Trumpp A, Beermann F. Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res. 2005;65:4005–4011. - PubMed
-
- Bennett DC. Genetics of melanoma progression: the rise and fall of cell senescence. Pigment Cell Melanoma Res. 2016;29:122–140. - PubMed
-
- Besch R, Berking C. POU transcription factors in melanocytes and melanoma. Eur J Cell Biol. 2014;93:55–60. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
