

Exposure of Barrett's and esophageal adenocarcinoma cells to bile acids activates EGFR-STAT3 signaling axis via induction of APE1
- PMID: 29991802
- PMCID: PMC6328352
- DOI: 10.1038/s41388-018-0388-8
Exposure of Barrett's and esophageal adenocarcinoma cells to bile acids activates EGFR-STAT3 signaling axis via induction of APE1
Abstract
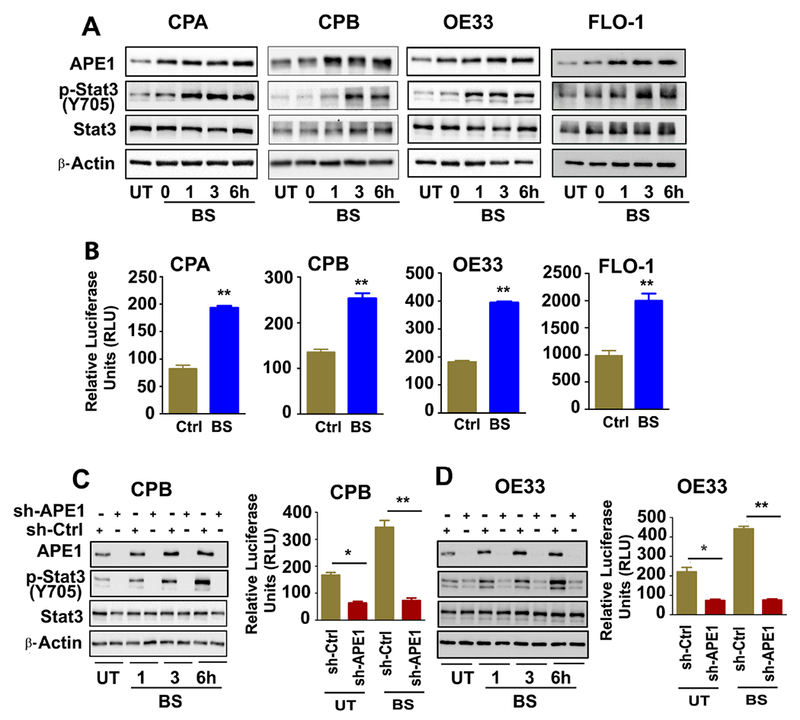
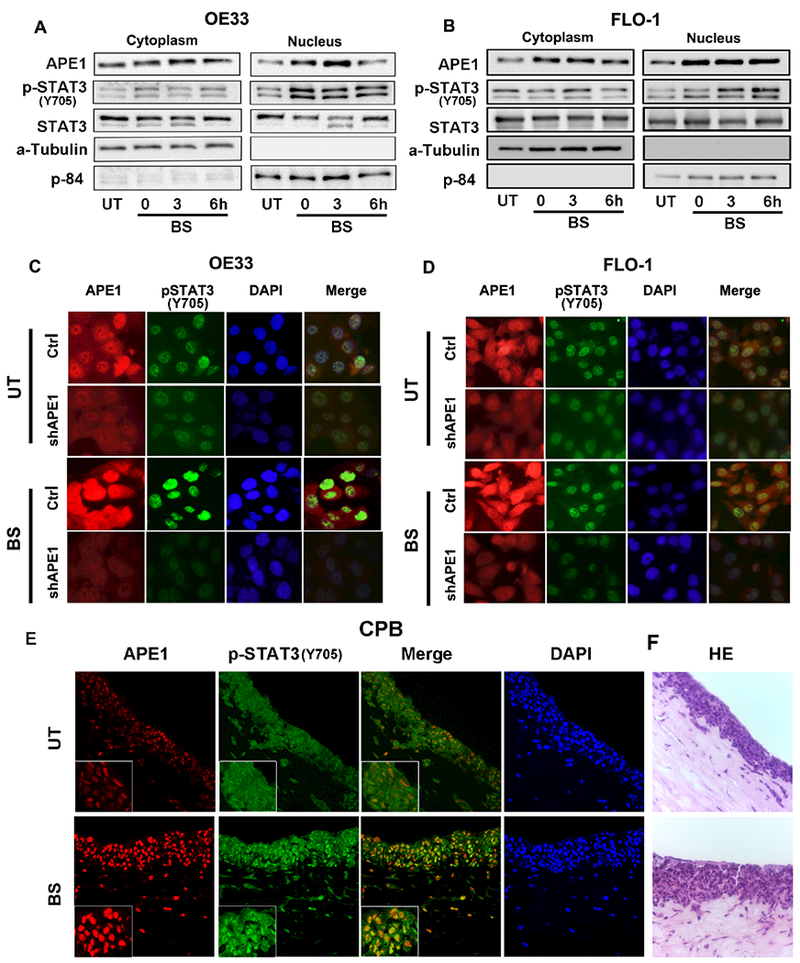
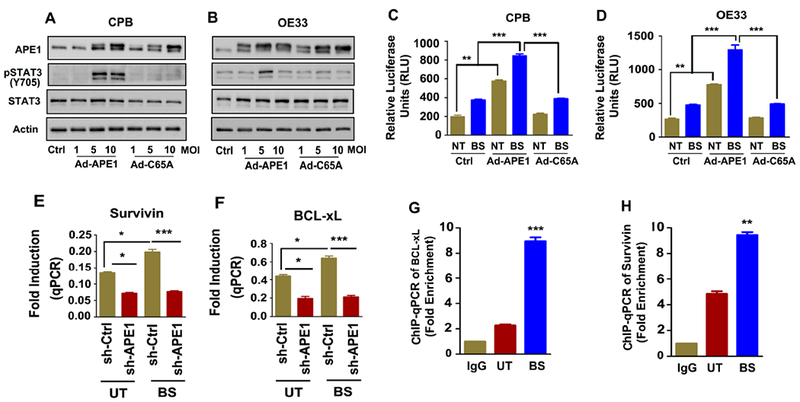
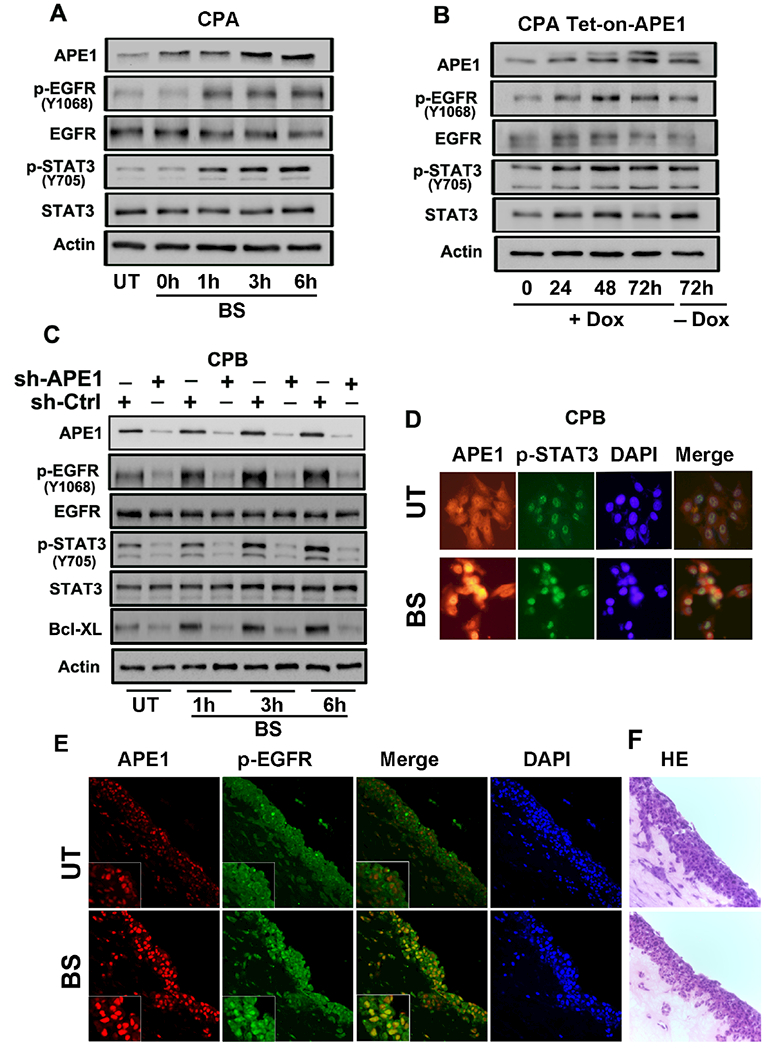
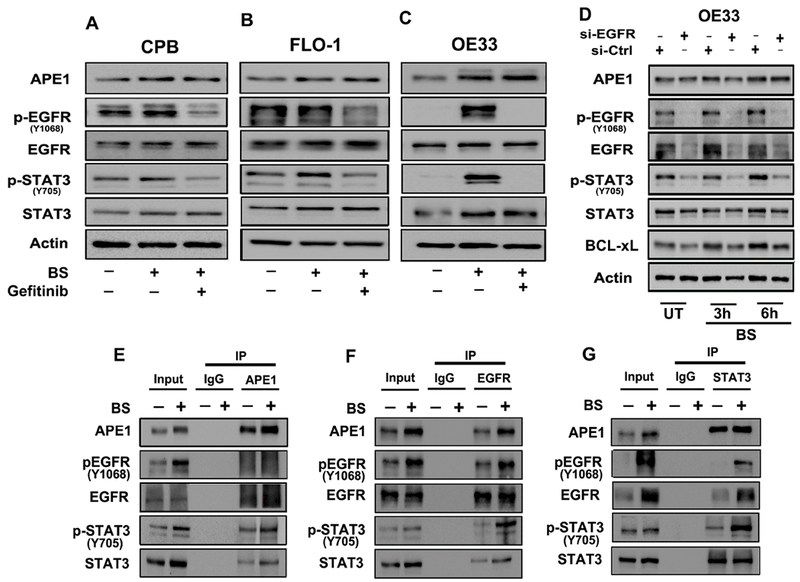
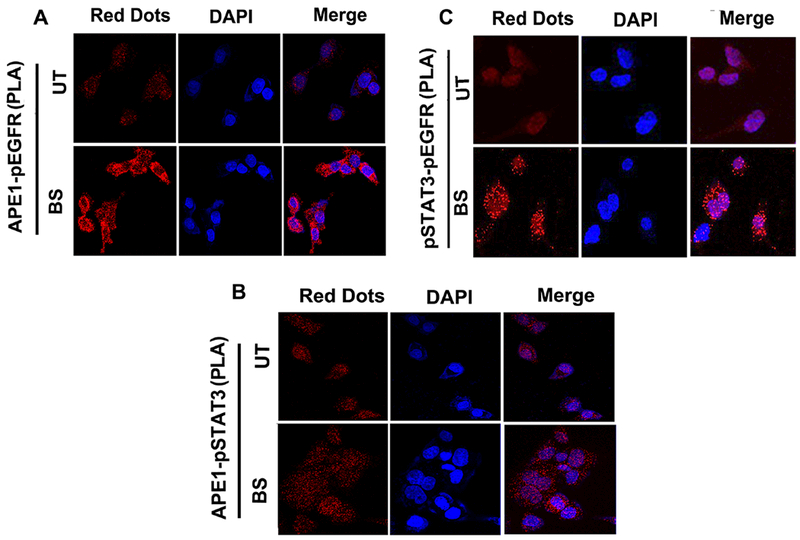
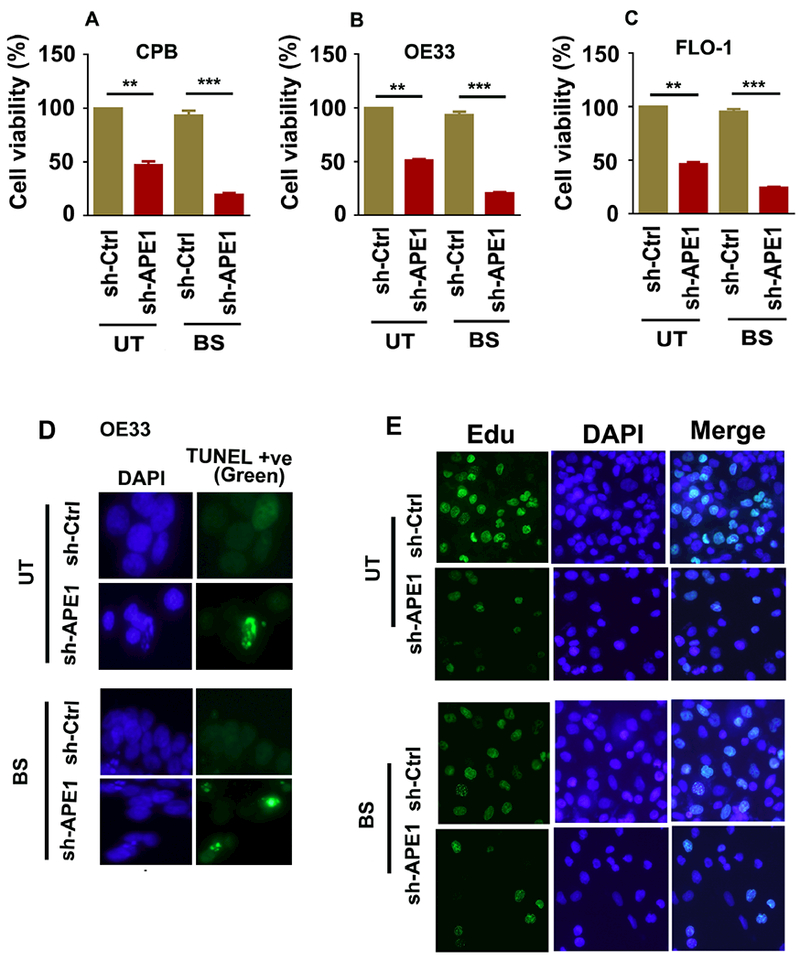
The development of Barrett's esophagus (BE) and its progression to esophageal adenocarcinoma (EAC) is highly linked to exposure to acidic bile salts due to chronic gastroesophageal reflux disease (GERD). In this study, we investigated the role of Apurinic/apyrimidinic endonuclease 1/redox effector factor-1 (APE1/REF-1) in STAT3 activation in response to acidic bile salts. Our results indicate that APE1 is constitutively overexpressed in EAC, whereas its expression is transiently induced in response to acidic bile salts in non-neoplastic BE. Using overexpression or shRNA knockdown of APE1, we found that APE1 is required for phosphorylation, nuclear localization, and transcriptional activation of STAT3. By using an APE1 redox-specific mutant (C65A) and APE1 redox inhibitor (E3330), we demonstrate that APE1 activates STAT3 in a redox-dependent manner. By using pharmacologic inhibitors and genetic knockdown systems, we found that EGFR is a required link between APE1 and STAT3. EGFR phosphorylation (Y1068) was directly associated with APE1 levels and redox function. Co-immunoprecipitation and proximity ligation assays indicated that APE1 coexists and interacts with the EGFR-STAT3 protein complex. Consistent with these findings, we demonstrated a significant induction in mRNA expression levels of STAT3 target genes (IL-6, IL-17A, BCL-xL, Survivin, and c-MYC) in BE and EAC cells, following acidic bile salts treatment. ChIP assays indicated that acidic bile salts treatment enhances binding of STAT3 to the promoter of its target genes, Survivin and BCL-xL. Inhibition of APE1/REF-1 redox activity using E3330 abrogated STAT3 DNA binding and transcriptional activity. The induction of APE1-STAT3 axis in acidic bile salts conditions provided a survival advantage and promoted cellular proliferation. In summary, our study provides multiple pieces of evidence supporting a critical role for APE1 induction in activating the EGFR-STAT3 signaling axis in response to acidic bile salts, the main risk factor for Barrett's carcinogenesis.
Conflict of interest statement
Figures
References
-
- Eloubeidi MA, Mason AC, Desmond RA, El-Serag HB. Temporal trends (1973-1997) in survival of patients with esophageal adenocarcinoma in the United States: a glimmer of hope? Am J Gastroenterol. 2003;98(7):1627–33. - PubMed
-
- Pera M, Manterola C, Vidal O, Grande L. Epidemiology of esophageal adenocarcinoma. J Surg Oncol. 2005;92(3):151–9. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- 1IK6BX003787/U.S. Department of Veterans Affairs (VA)/International
- R01 CA138833/CA/NCI NIH HHS/United States
- R01 CA206564/CA/NCI NIH HHS/United States
- R01 CA206563/CA/NCI NIH HHS/United States
- R01CA206563/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
