

Epigenetic regulation in development: is the mouse a good model for the human?
- PMID: 29992283
- PMCID: PMC6093373
- DOI: 10.1093/humupd/dmy021
Epigenetic regulation in development: is the mouse a good model for the human?
Abstract
Background: Over the past few years, advances in molecular technologies have allowed unprecedented mapping of epigenetic modifications in gametes and during early embryonic development. This work is allowing a detailed genomic analysis, which for the first time can answer long-standing questions about epigenetic regulation and reprogramming, and highlights differences between mouse and human, the implications of which are only beginning to be explored.
Objective and rationale: In this review, we summarise new low-cell molecular methods enabling the interrogation of epigenetic information in gametes and early embryos, the mechanistic insights these have provided, and contrast the findings in mouse and human.
Search methods: Relevant studies were identified by PubMed search.
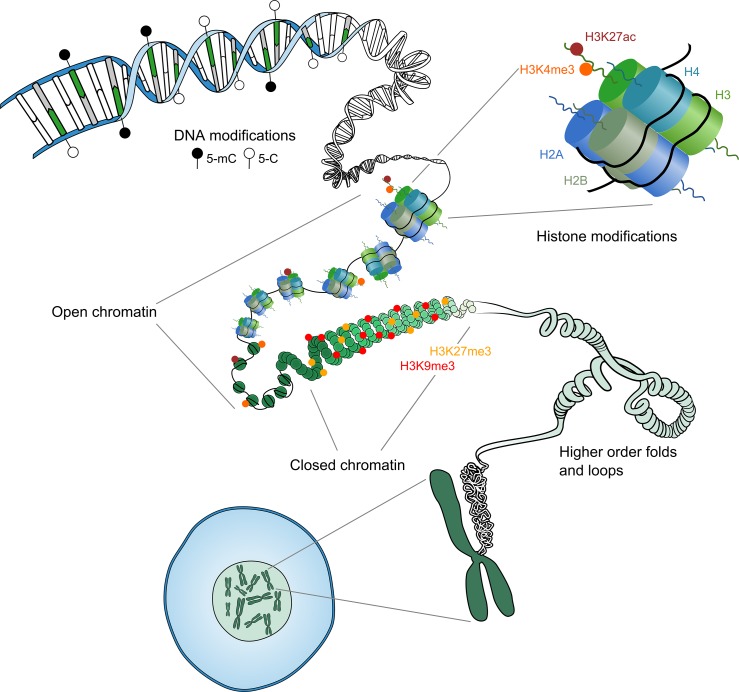
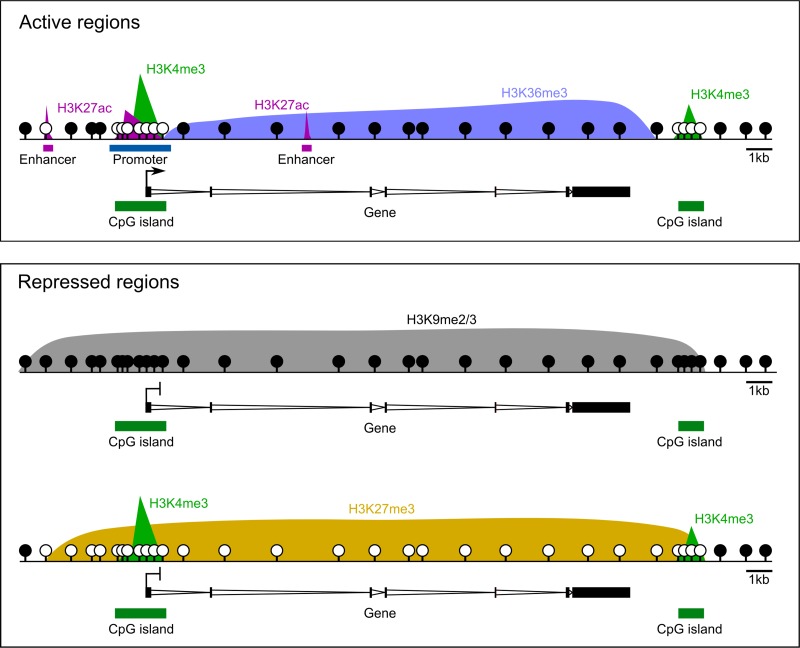
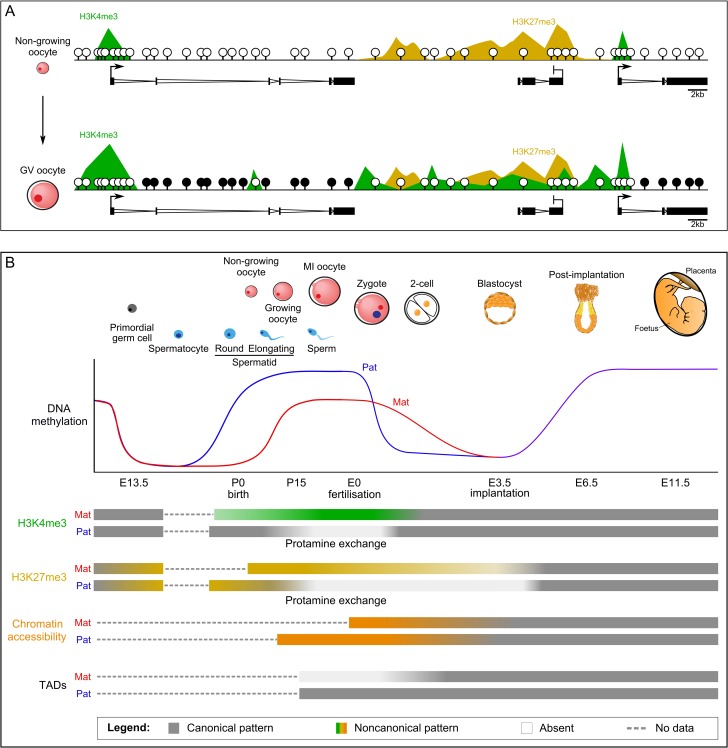
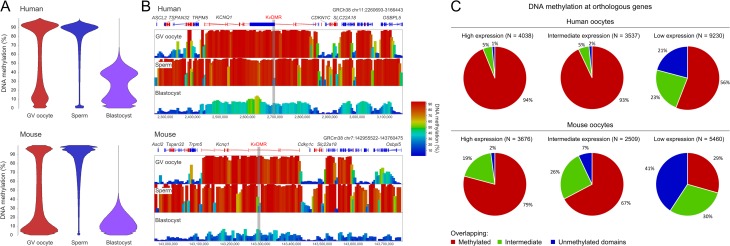
Outcomes: We discuss the levels of epigenetic regulation, from DNA modifications to chromatin organisation, during mouse gametogenesis, fertilisation and pre- and post-implantation development. The recently characterised features of the oocyte epigenome highlight its exceptionally unique regulatory landscape. The chromatin organisation and epigenetic landscape of both gametic genomes are rapidly reprogrammed after fertilisation. This extensive epigenetic remodelling is necessary for zygotic genome activation, but the mechanistic link remains unclear. While the vast majority of epigenetic information from the gametes is erased in pre-implantation development, new insights suggest that repressive histone modifications from the oocyte may mediate a novel mechanism of imprinting. To date, the characterisation of epigenetics in human development has been almost exclusively limited to DNA methylation profiling; these data reinforce that the global dynamics are conserved between mouse and human. However, as we look closer, it is becoming apparent that the mechanisms regulating these dynamics are distinct. These early findings emphasise the importance of investigations of fundamental epigenetic mechanisms in both mouse and humans.
Wider implications: Failures in epigenetic regulation have been implicated in human disease and infertility. With increasing maternal age and use of reproductive technologies in countries all over the world, it is becoming ever more important to understand the necessary processes required to establish a developmentally competent embryo. Furthermore, it is essential to evaluate the extent to which these epigenetic patterns are sensitive to such technologies and other adverse environmental exposures.
Figures
References
-
- Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet 2016;8:487–500. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
