

Reprogramming human T cell function and specificity with non-viral genome targeting
- PMID: 29995861
- PMCID: PMC6239417
- DOI: 10.1038/s41586-018-0326-5
Reprogramming human T cell function and specificity with non-viral genome targeting
Abstract
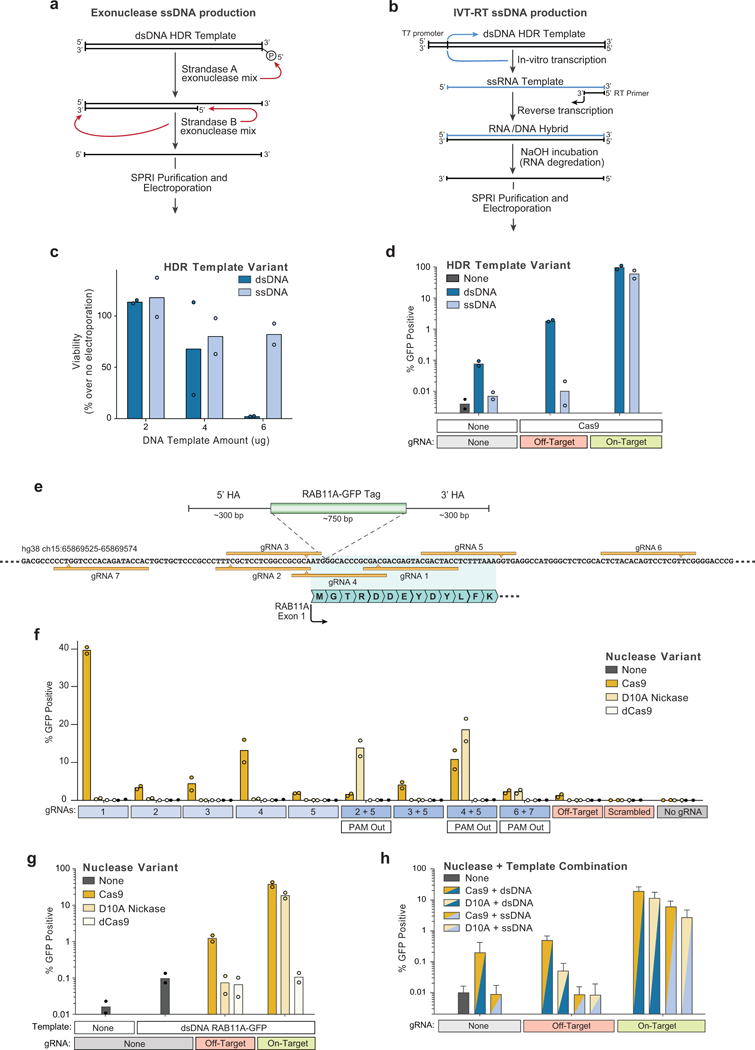
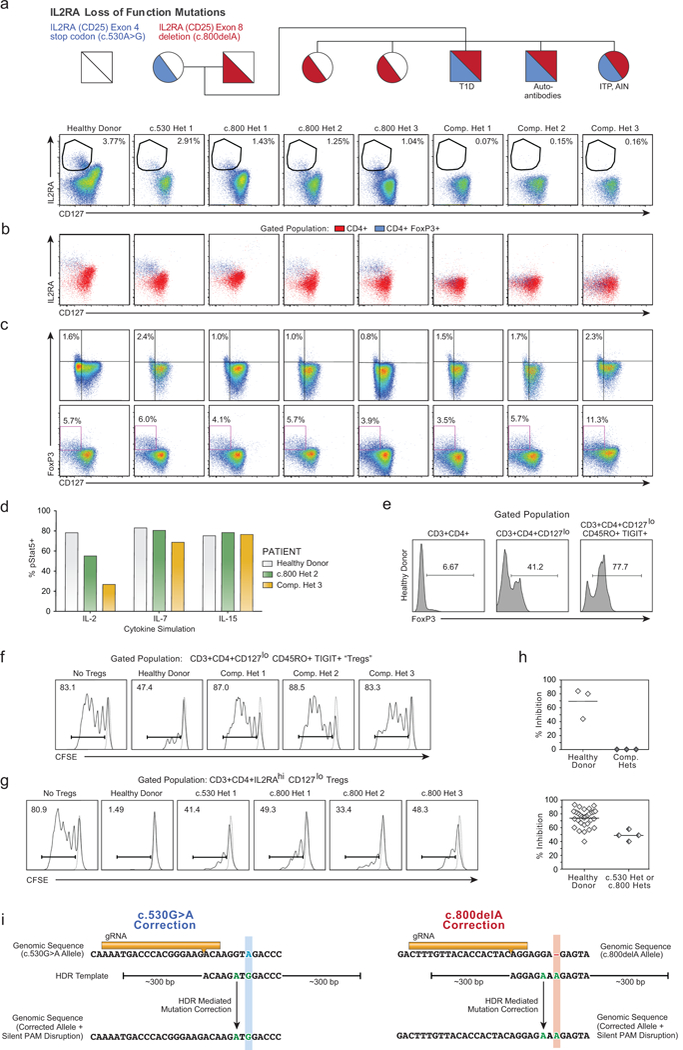
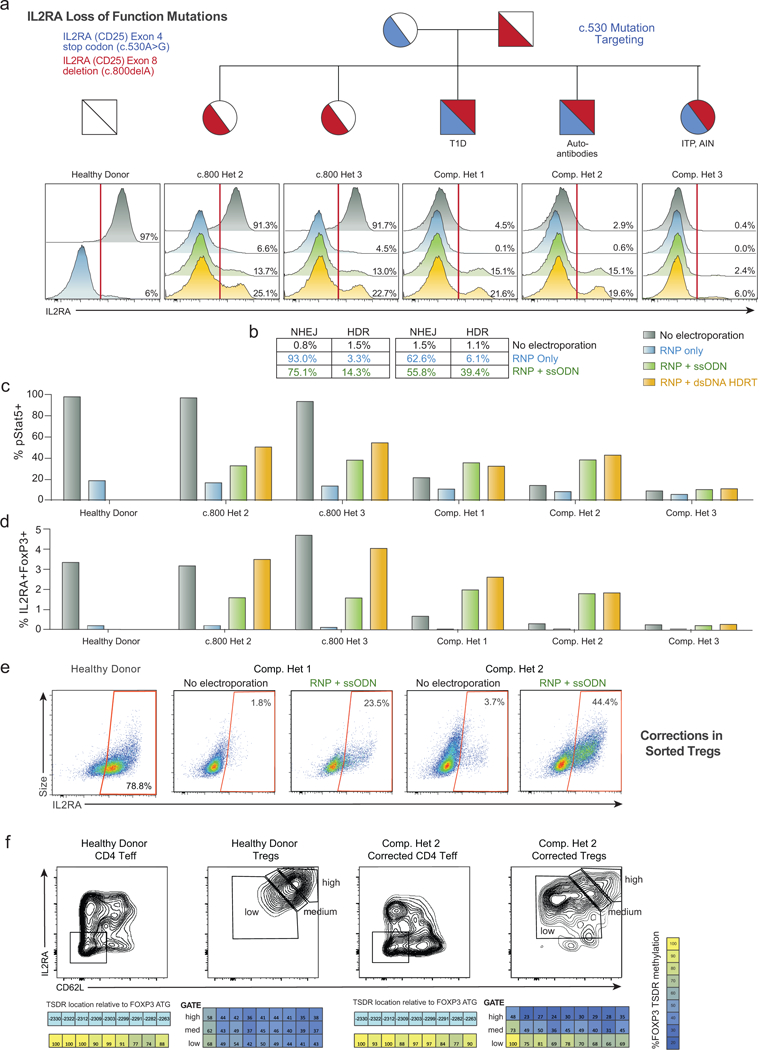
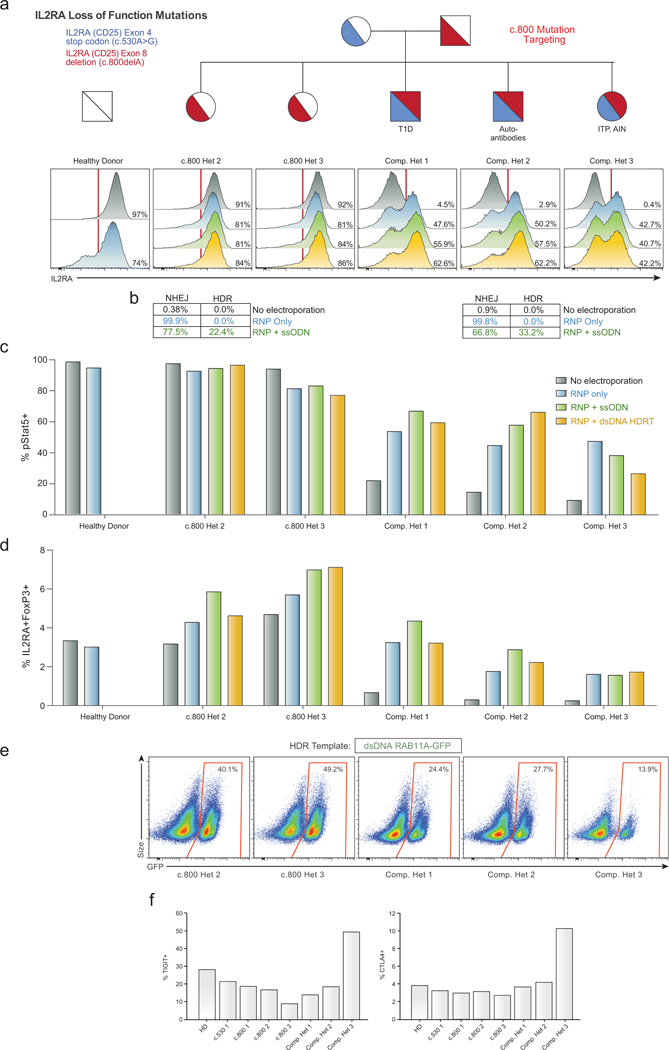
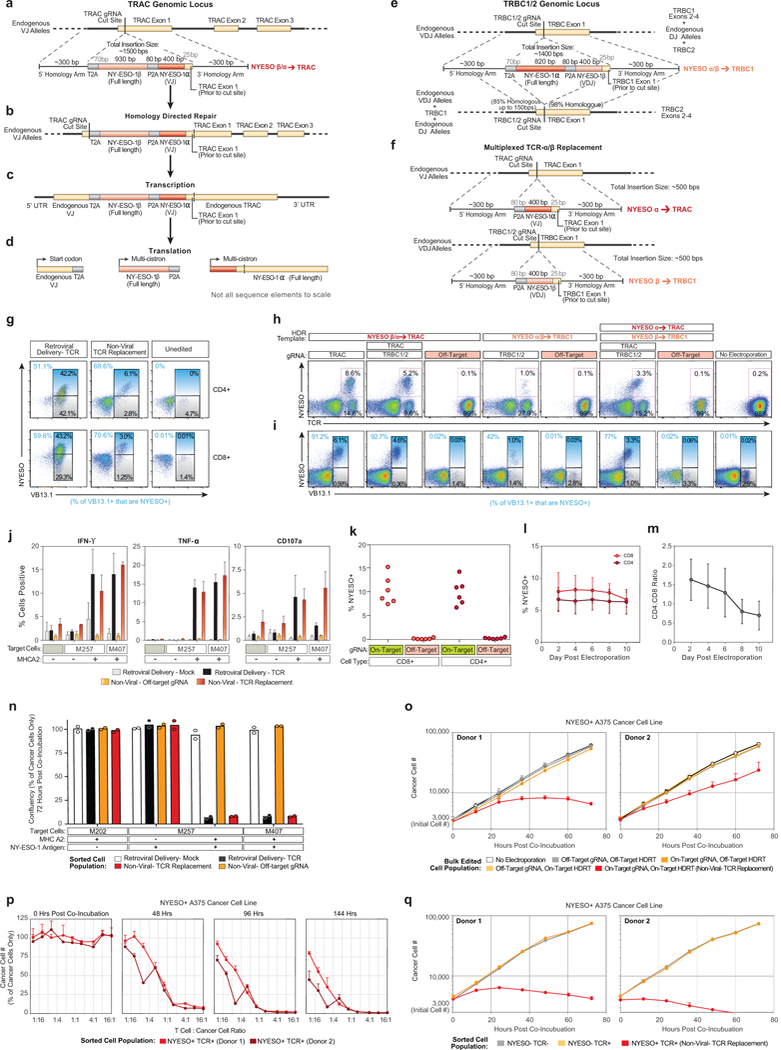
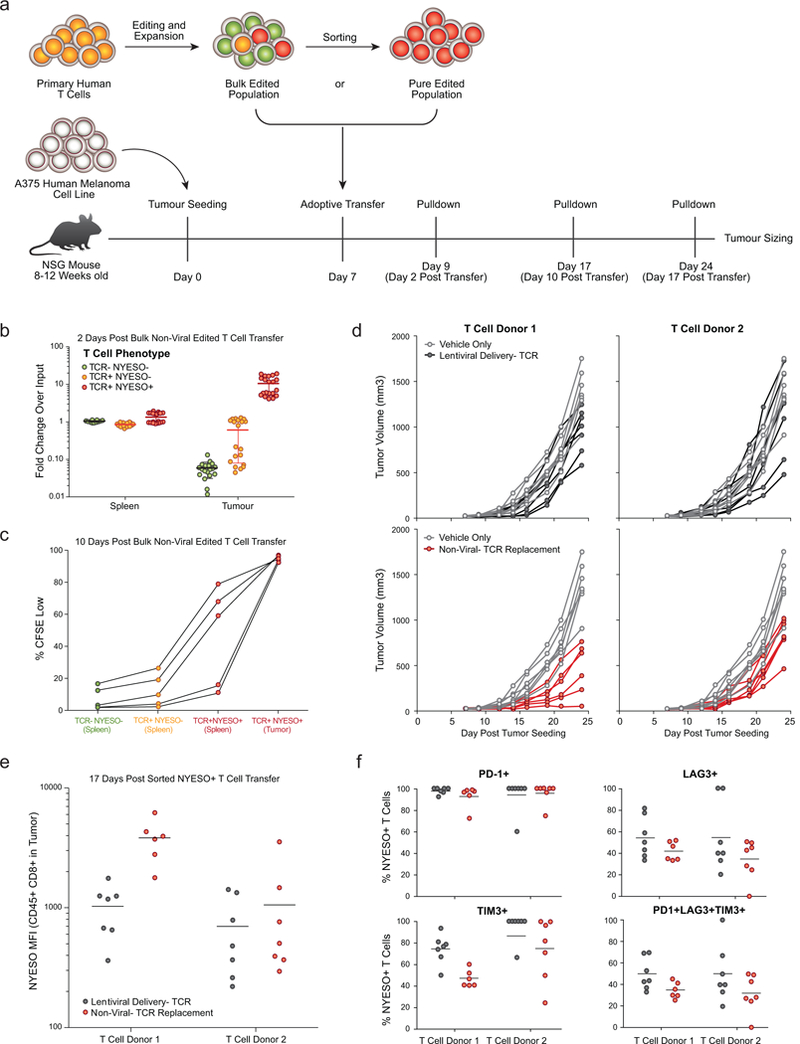
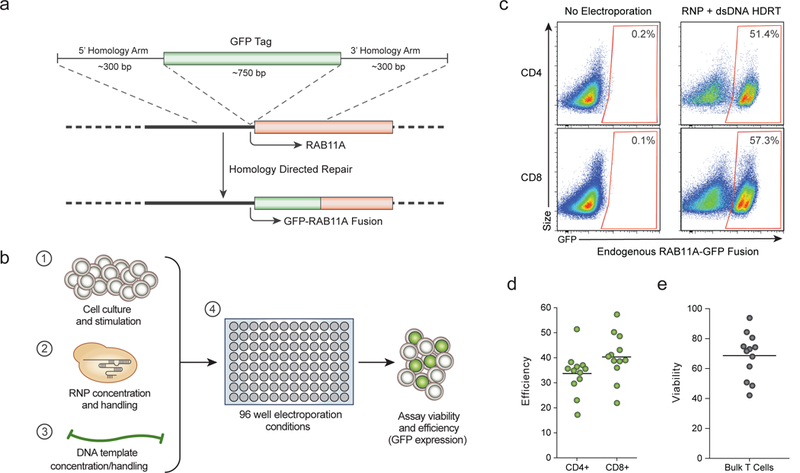
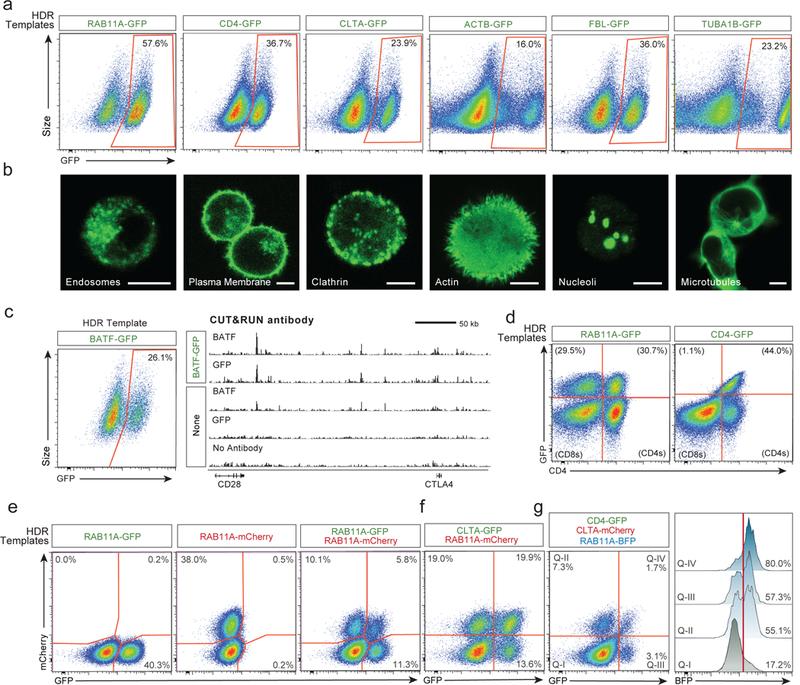
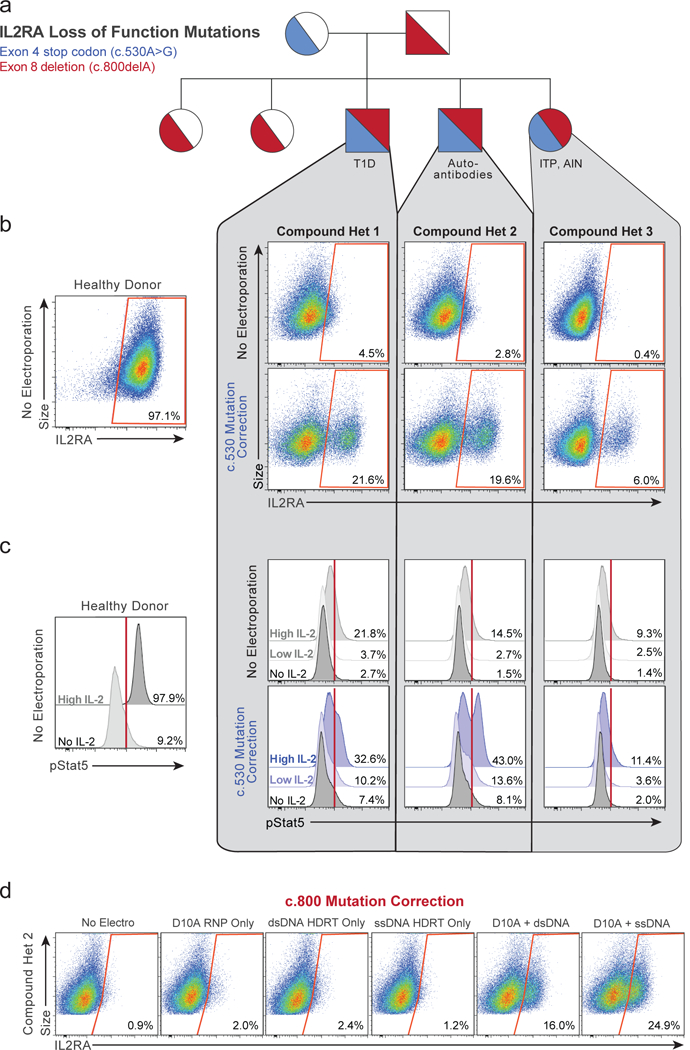
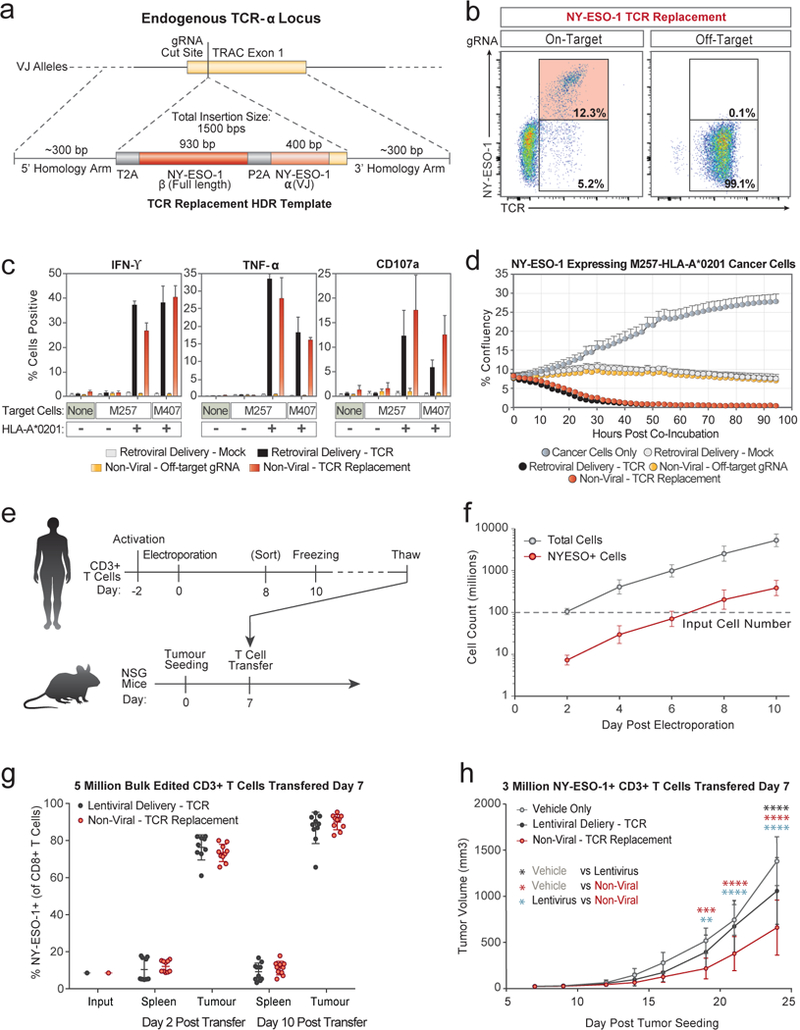
Decades of work have aimed to genetically reprogram T cells for therapeutic purposes1,2 using recombinant viral vectors, which do not target transgenes to specific genomic sites3,4. The need for viral vectors has slowed down research and clinical use as their manufacturing and testing is lengthy and expensive. Genome editing brought the promise of specific and efficient insertion of large transgenes into target cells using homology-directed repair5,6. Here we developed a CRISPR-Cas9 genome-targeting system that does not require viral vectors, allowing rapid and efficient insertion of large DNA sequences (greater than one kilobase) at specific sites in the genomes of primary human T cells, while preserving cell viability and function. This permits individual or multiplexed modification of endogenous genes. First, we applied this strategy to correct a pathogenic IL2RA mutation in cells from patients with monogenic autoimmune disease, and demonstrate improved signalling function. Second, we replaced the endogenous T cell receptor (TCR) locus with a new TCR that redirected T cells to a cancer antigen. The resulting TCR-engineered T cells specifically recognized tumour antigens and mounted productive anti-tumour cell responses in vitro and in vivo. Together, these studies provide preclinical evidence that non-viral genome targeting can enable rapid and flexible experimental manipulation and therapeutic engineering of primary human immune cells.
Figures
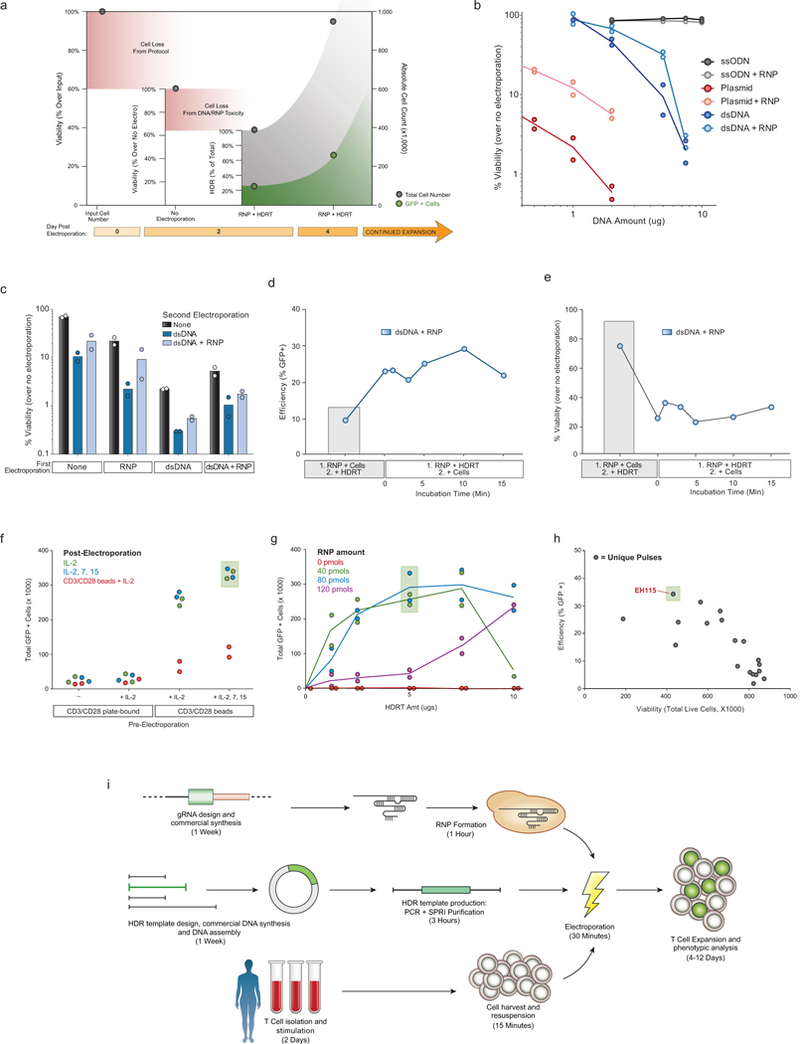
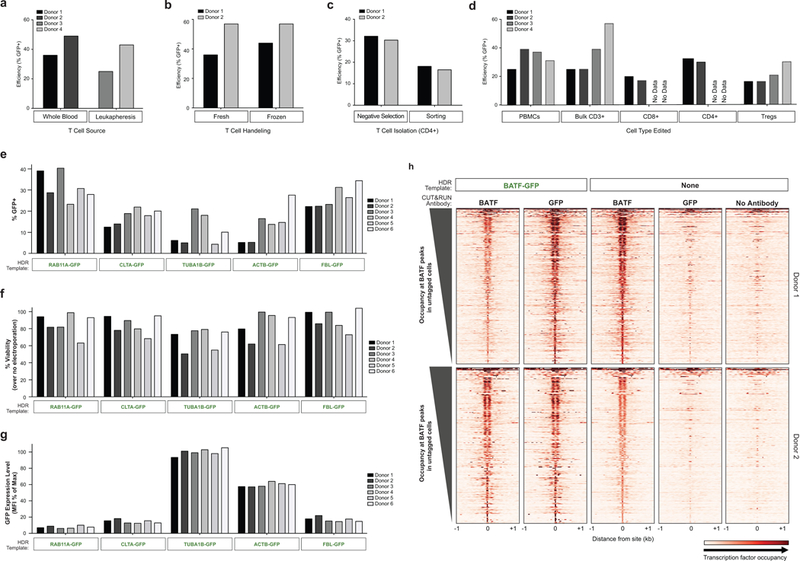
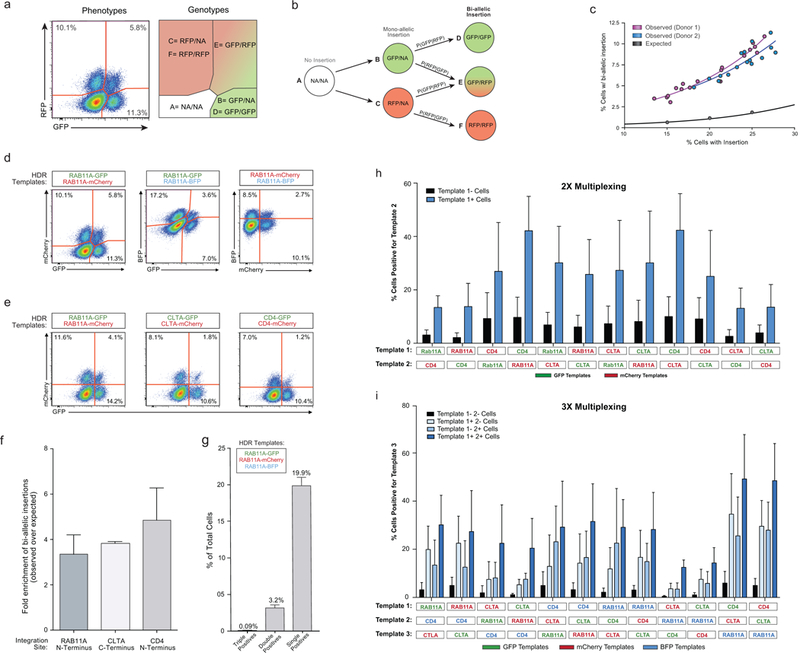
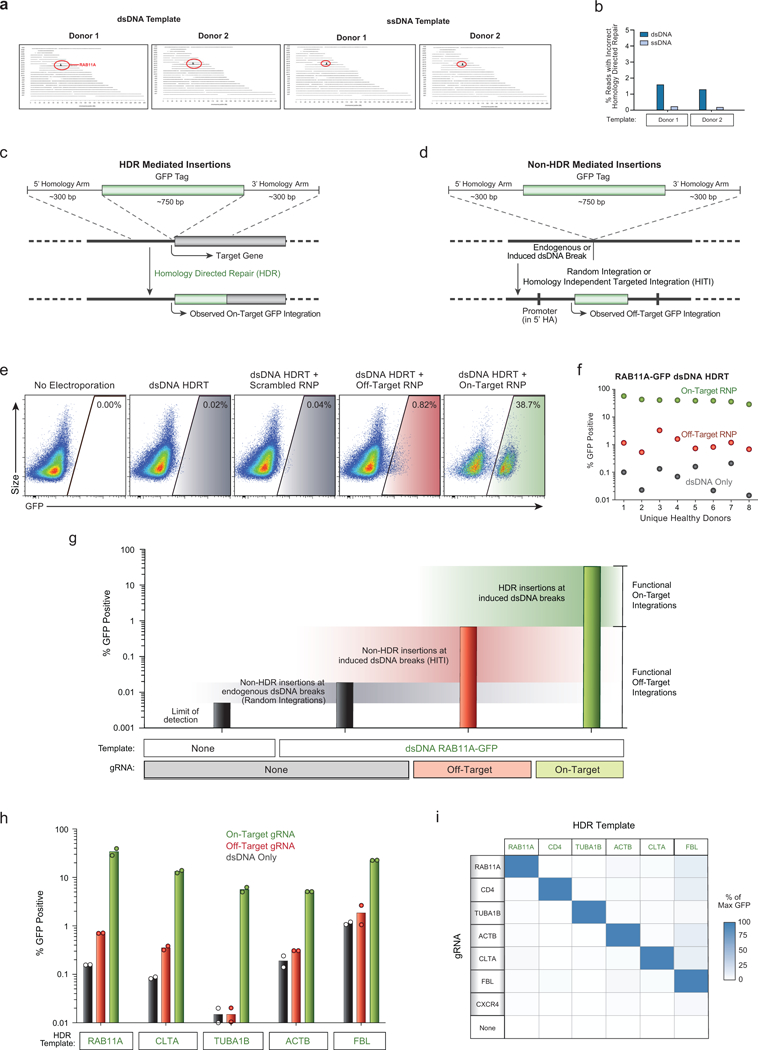
References
-
- Verhoeyen E, Costa C & Cosset F-L Lentiviral vector gene transfer into human T cells. Methods Mol. Biol. 506, 97–114 (2009). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- T32 GM007618/GM/NIGMS NIH HHS/United States
- L40 AI140341/AI/NIAID NIH HHS/United States
- P30 CA082103/CA/NCI NIH HHS/United States
- R35 CA197633/CA/NCI NIH HHS/United States
- S10 OD021822/OD/NIH HHS/United States
- T32 AI007641/AI/NIAID NIH HHS/United States
- P50 GM082250/GM/NIGMS NIH HHS/United States
- T32 CA108462/CA/NCI NIH HHS/United States
- K23 DK094866/DK/NIDDK NIH HHS/United States
- K23 AI115001/AI/NIAID NIH HHS/United States
- P30 DK020595/DK/NIDDK NIH HHS/United States
- P30 DK063720/DK/NIDDK NIH HHS/United States
- DP3 DK111914/DK/NIDDK NIH HHS/United States
- T32 AI007334/AI/NIAID NIH HHS/United States
- T32 DK007418/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
