

Estrogen promotes estrogen receptor negative BRCA1-deficient tumor initiation and progression
- PMID: 29996906
- PMCID: PMC6042319
- DOI: 10.1186/s13058-018-0996-9
Estrogen promotes estrogen receptor negative BRCA1-deficient tumor initiation and progression
Abstract
Background: Estrogen promotes breast cancer development and progression mainly through estrogen receptor (ER). However, blockage of estrogen production or action prevents development of and suppresses progression of ER-negative breast cancers. How estrogen promotes ER-negative breast cancer development and progression is poorly understood. We previously discovered that deletion of cell cycle inhibitors p16Ink4a (p16) or p18Ink4c (p18) is required for development of Brca1-deficient basal-like mammary tumors, and that mice lacking p18 develop luminal-type mammary tumors.
Methods: A genetic model system with three mouse strains, one that develops ER-positive mammary tumors (p18 single deletion) and the others that develop ER-negative tumors (p16;Brca1 and p18;Brca1 compound deletion), human BRCA1 mutant breast cancer patient-derived xenografts, and human BRCA1-deficient and BRCA1-proficient breast cancer cells were used to determine the role of estrogen in activating epithelial-mesenchymal transition (EMT), stimulating cell proliferation, and promoting ER-negative mammary tumor initiation and metastasis.
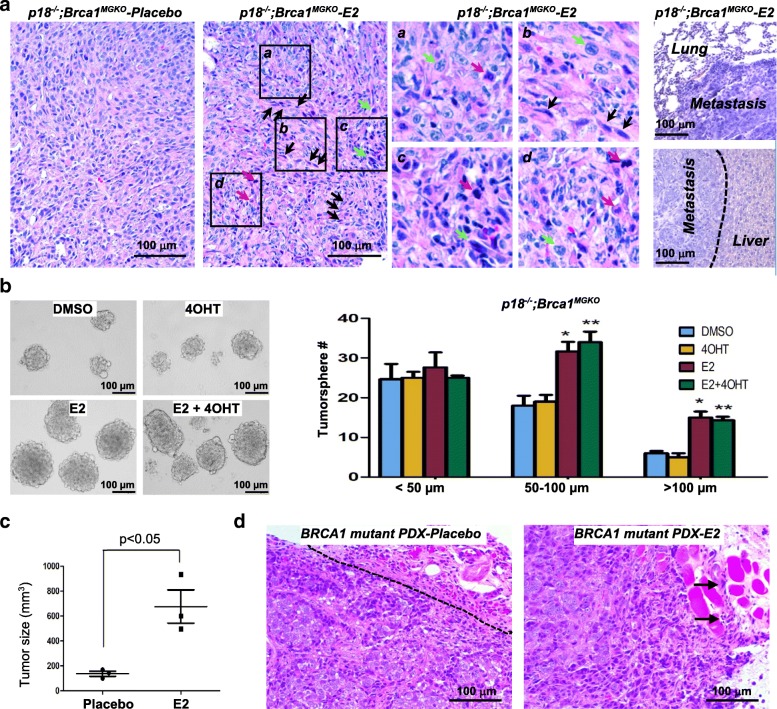
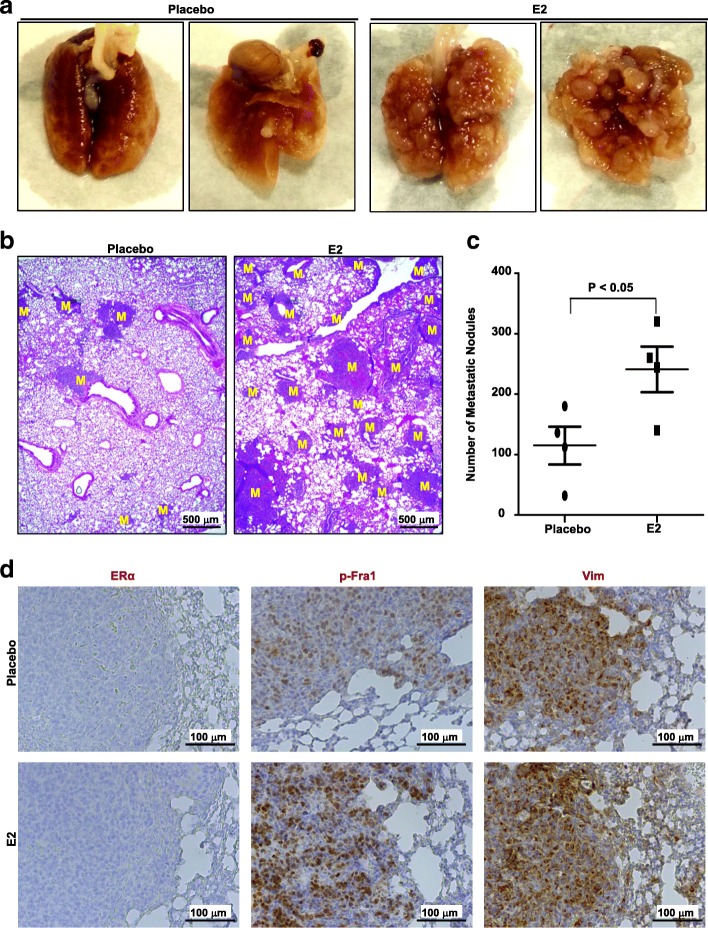
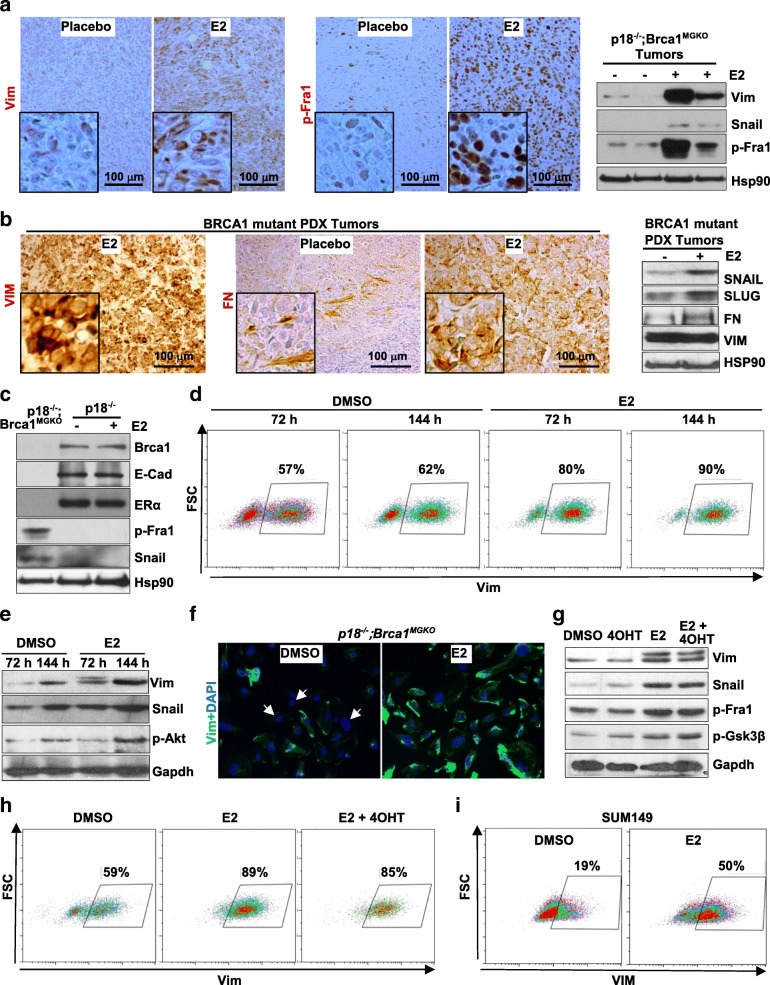
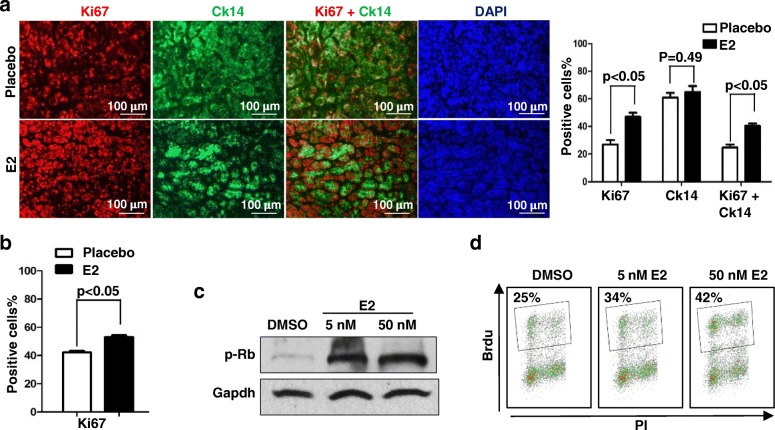
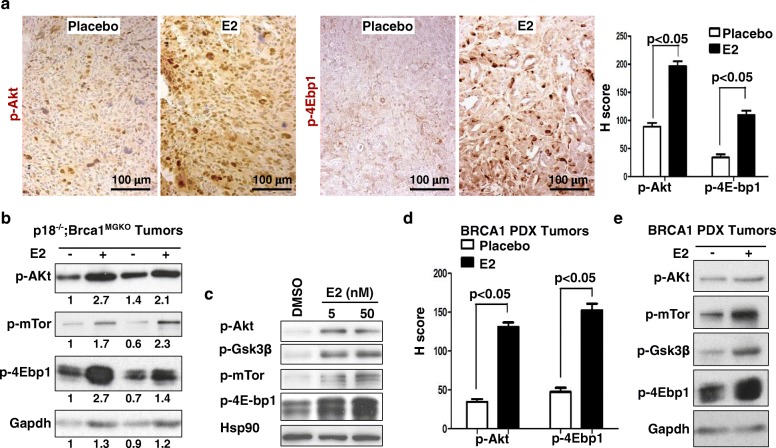
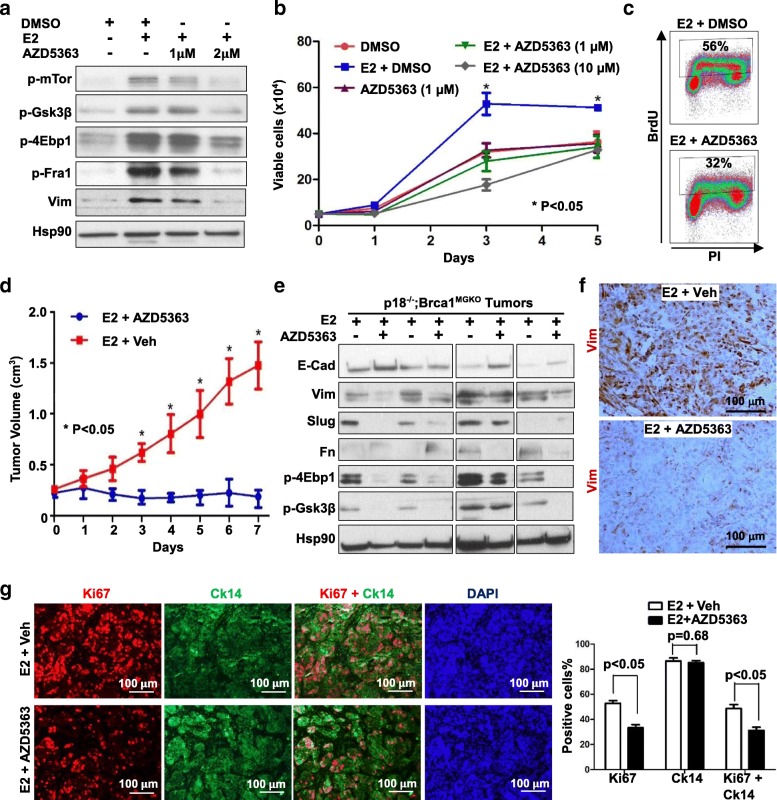
Results: Estrogen stimulated the proliferation and tumor-initiating potential of both ER-positive Brca1-proficient and ER-negative Brca1-deficient tumor cells. Estrogen activated EMT in a subset of Brca1-deficient mammary tumor cells that maintained epithelial features, and enhanced the number of cancer stem cells, promoting tumor progression and metastasis. Estrogen activated EMT independent of ER in Brca1-deficient, but not Brca1-proficient, tumor cells. Estrogen activated the AKT pathway in BRCA1-deficient tumor cells independent of ER, and pharmaceutical inhibition of AKT activity suppressed EMT and cell proliferation preventing BRCA1 deficient tumor progression.
Conclusions: This study reveals for the first time that estrogen promotes BRCA1-deficient tumor initiation and progression by stimulation of cell proliferation and activation of EMT, which are dependent on AKT activation and independent of ER.
Keywords: BRCA1; Cancer stem cells; EMT; Estrogen; Estrogen receptor.
Conflict of interest statement
Ethics approval
The Institutional Animal Care and Use Committee at the University of Miami approved all animal procedures.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
GATA3 functions downstream of BRCA1 to suppress EMT in breast cancer.Theranostics. 2021 Jul 13;11(17):8218-8233. doi: 10.7150/thno.59280. eCollection 2021. Theranostics. 2021. PMID: 34373738 Free PMC article.
-
PDGFRβ is an essential therapeutic target for BRCA1-deficient mammary tumors.Breast Cancer Res. 2021 Jan 21;23(1):10. doi: 10.1186/s13058-021-01387-x. Breast Cancer Res. 2021. PMID: 33478572 Free PMC article.
-
BRCA1 suppresses epithelial-to-mesenchymal transition and stem cell dedifferentiation during mammary and tumor development.Cancer Res. 2014 Nov 1;74(21):6161-72. doi: 10.1158/0008-5472.CAN-14-1119. Epub 2014 Sep 19. Cancer Res. 2014. PMID: 25239453
-
Activating Mutations of ESR1, BRCA1 and CYP19 Aromatase Genes Confer Tumor Response in Breast Cancers Treated with Antiestrogens.Recent Pat Anticancer Drug Discov. 2017;12(2):136-147. doi: 10.2174/1574892812666170227110842. Recent Pat Anticancer Drug Discov. 2017. PMID: 28245776 Review.
-
BRCA1 and estrogen/estrogen receptor in breast cancer: where they interact?Int J Biol Sci. 2014 May 14;10(5):566-75. doi: 10.7150/ijbs.8579. eCollection 2014. Int J Biol Sci. 2014. PMID: 24910535 Free PMC article. Review.
Cited by
-
GATA3 functions downstream of BRCA1 to promote DNA damage repair and suppress dedifferentiation in breast cancer.BMC Biol. 2024 Apr 16;22(1):85. doi: 10.1186/s12915-024-01881-6. BMC Biol. 2024. PMID: 38627785 Free PMC article.
-
Proteomic Markers for Mechanobiological Properties of Metastatic Cancer Cells.Int J Mol Sci. 2023 Mar 1;24(5):4773. doi: 10.3390/ijms24054773. Int J Mol Sci. 2023. PMID: 36902201 Free PMC article. Review.
-
GATA3 functions downstream of BRCA1 to suppress EMT in breast cancer.Theranostics. 2021 Jul 13;11(17):8218-8233. doi: 10.7150/thno.59280. eCollection 2021. Theranostics. 2021. PMID: 34373738 Free PMC article.
-
Impaired histone inheritance promotes tumor progression.Nat Commun. 2023 Jun 10;14(1):3429. doi: 10.1038/s41467-023-39185-y. Nat Commun. 2023. PMID: 37301892 Free PMC article.
-
PDGFRβ is an essential therapeutic target for BRCA1-deficient mammary tumors.Breast Cancer Res. 2021 Jan 21;23(1):10. doi: 10.1186/s13058-021-01387-x. Breast Cancer Res. 2021. PMID: 33478572 Free PMC article.
References
-
- Swain SM. Tamoxifen for patients with estrogen receptor-negative breast cancer. J Clin Oncol. 2001;19:93S–97S. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
