

miR-196b target screen reveals mechanisms maintaining leukemia stemness with therapeutic potential
- PMID: 29997117
- PMCID: PMC6080909
- DOI: 10.1084/jem.20171312
miR-196b target screen reveals mechanisms maintaining leukemia stemness with therapeutic potential
Abstract
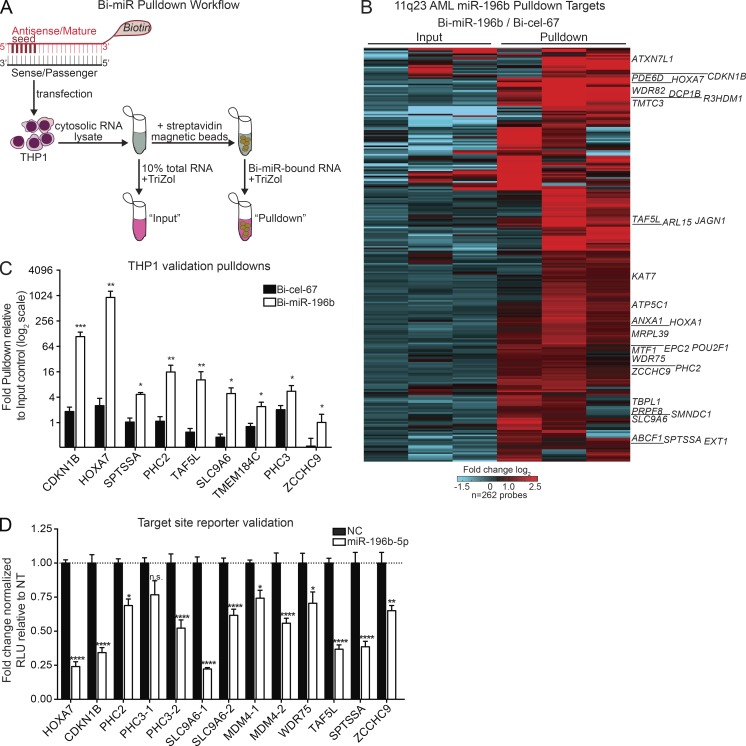
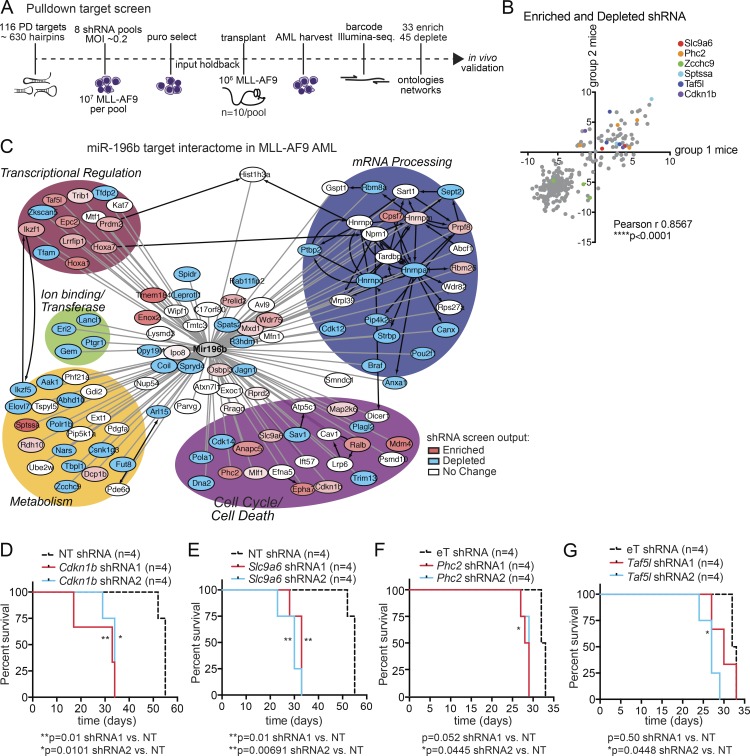
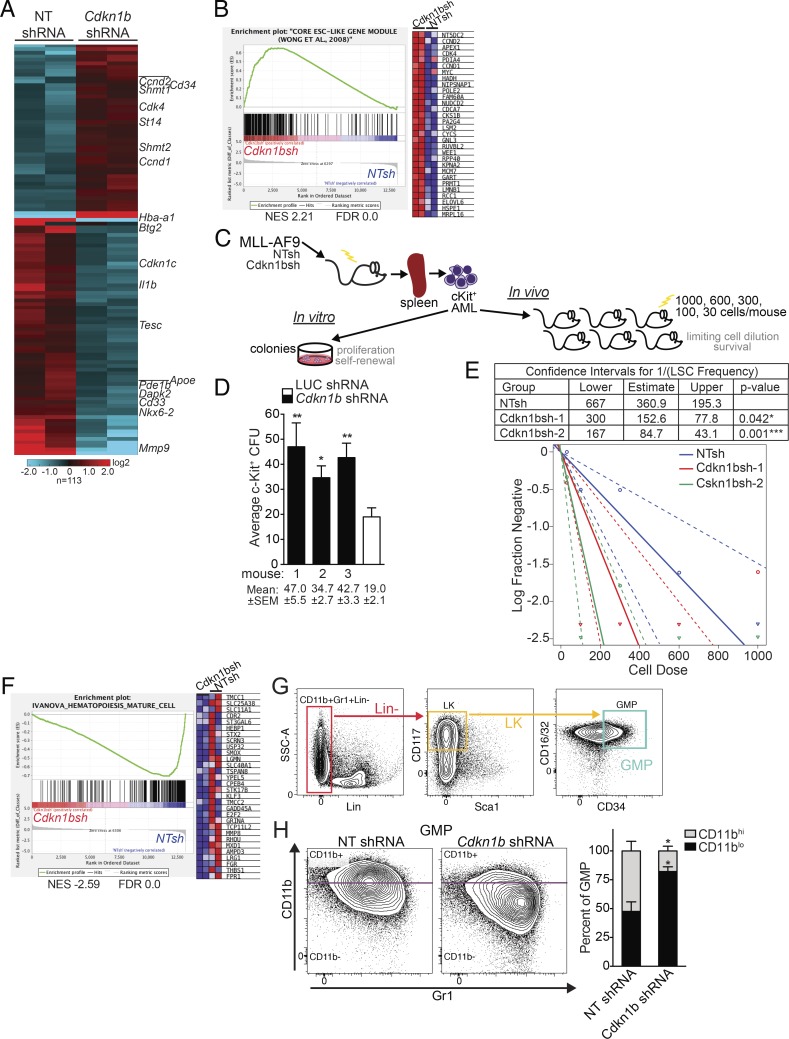
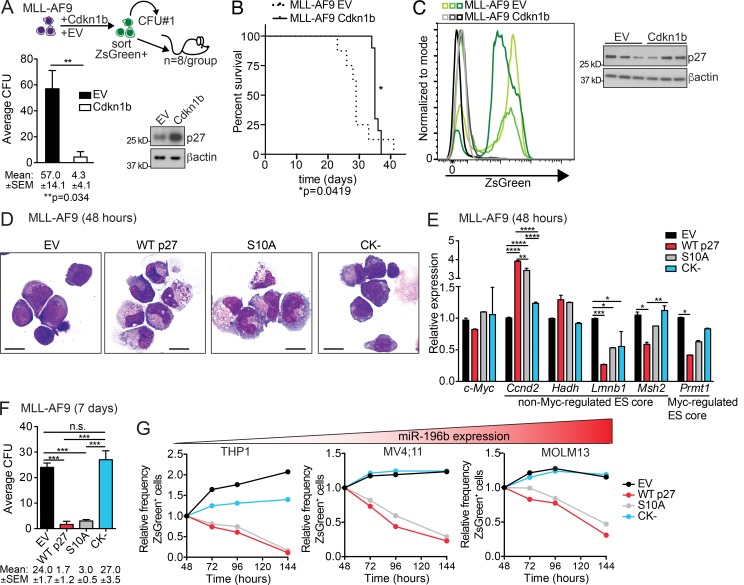
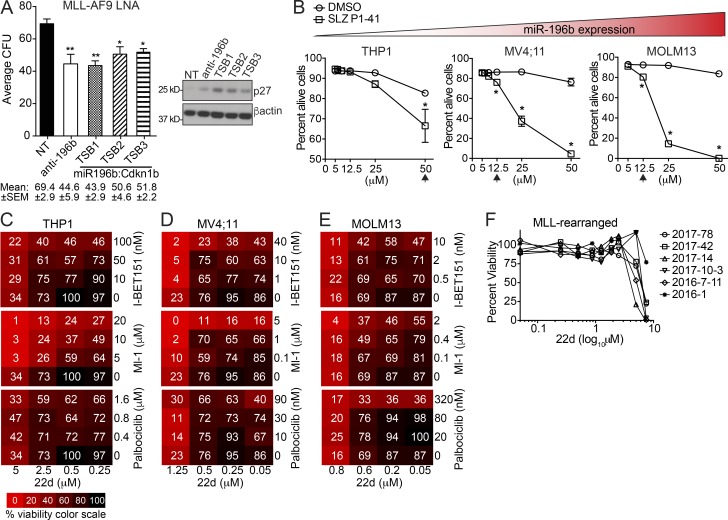
We have shown that antagomiR inhibition of miRNA miR-21 and miR-196b activity is sufficient to ablate MLL-AF9 leukemia stem cells (LSC) in vivo. Here, we used an shRNA screening approach to mimic miRNA activity on experimentally verified miR-196b targets to identify functionally important and therapeutically relevant pathways downstream of oncogenic miRNA in MLL-r AML. We found Cdkn1b (p27Kip1) is a direct miR-196b target whose repression enhanced an embryonic stem cell-like signature associated with decreased leukemia latency and increased numbers of leukemia stem cells in vivo. Conversely, elevation of p27Kip1 significantly reduced MLL-r leukemia self-renewal, promoted monocytic differentiation of leukemic blasts, and induced cell death. Antagonism of miR-196b activity or pharmacologic inhibition of the Cks1-Skp2-containing SCF E3-ubiquitin ligase complex increased p27Kip1 and inhibited human AML growth. This work illustrates that understanding oncogenic miRNA target pathways can identify actionable targets in leukemia.
© 2018 Crown copyright. The government of Australia, Canada, or the UK ("the Crown") owns the copyright interests of authors who are government employees. The Crown Copyright is not transferable.
Figures
References
-
- Armstrong S.A., Staunton J.E., Silverman L.B., Pieters R., den Boer M.L., Minden M.D., Sallan S.E., Lander E.S., Golub T.R., and Korsmeyer S.J.. 2002. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 30:41–47. 10.1038/ng765 - DOI - PubMed
-
- Betel D., Wilson M., Gabow A., Marks D.S., and Sander C.. 2008. The microRNA.org resource: targets and expression. Nucleic Acids Res. 36(Database):D149–D153. 10.1093/nar/gkm995 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
