

Therapeutic vulnerability of multiple myeloma to MIR17PTi, a first-in-class inhibitor of pri-miR-17-92
- PMID: 29997223
- PMCID: PMC6128086
- DOI: 10.1182/blood-2018-03-836601
Therapeutic vulnerability of multiple myeloma to MIR17PTi, a first-in-class inhibitor of pri-miR-17-92
Abstract
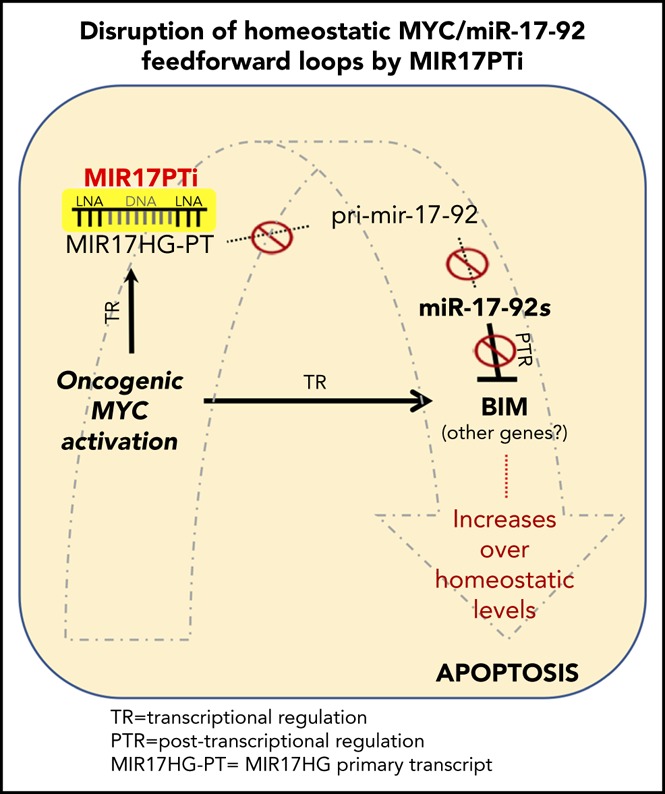
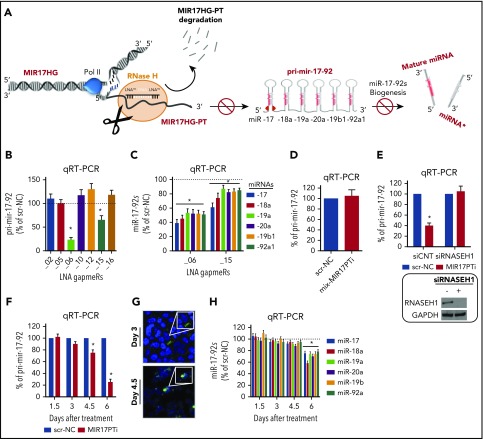
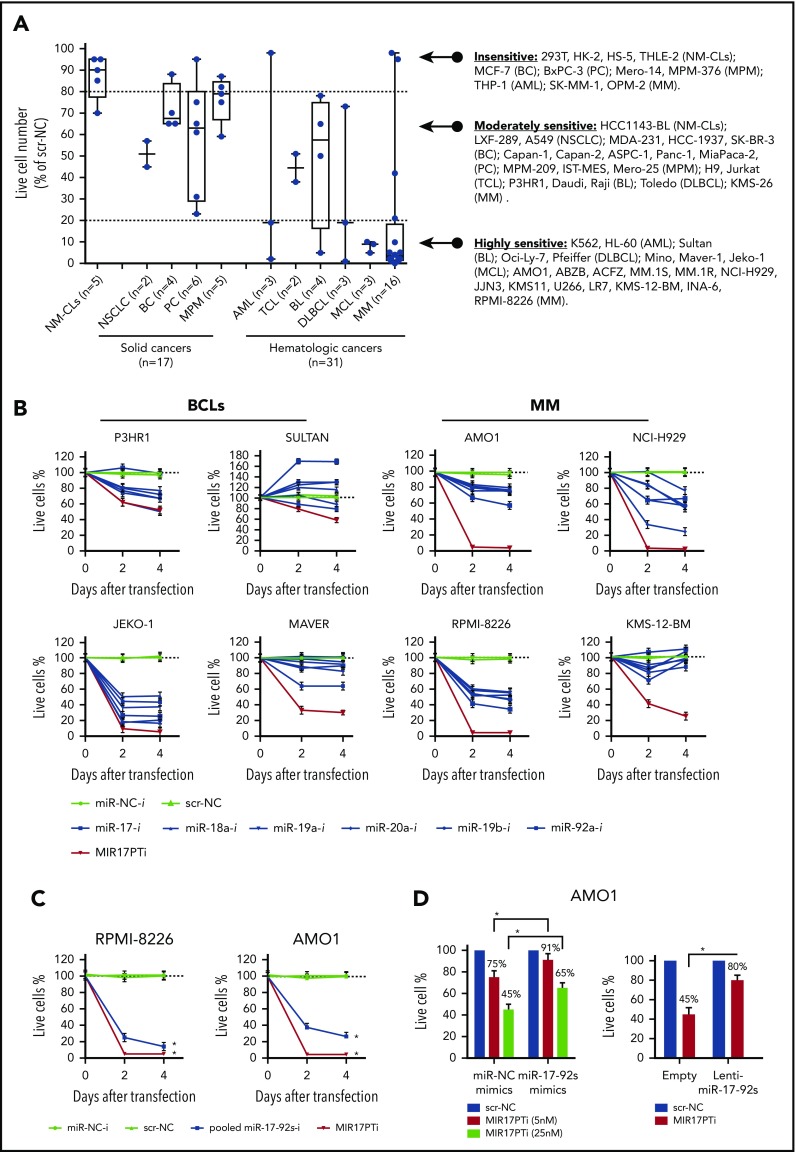
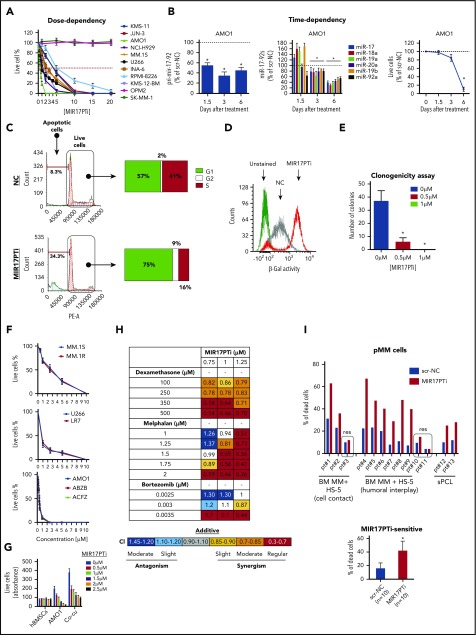
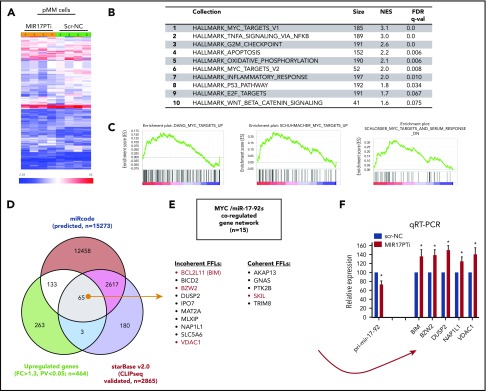
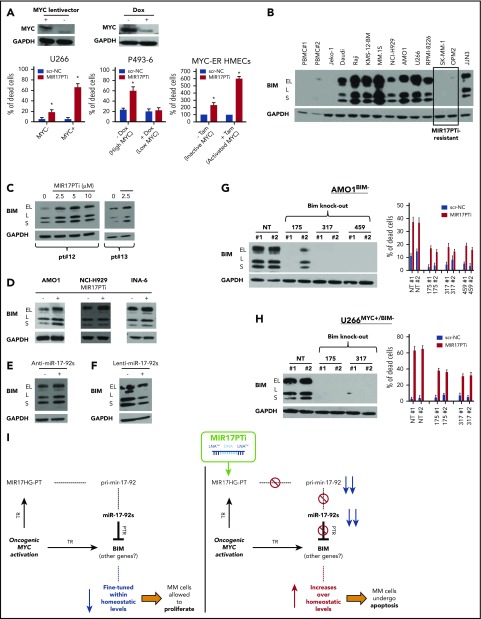
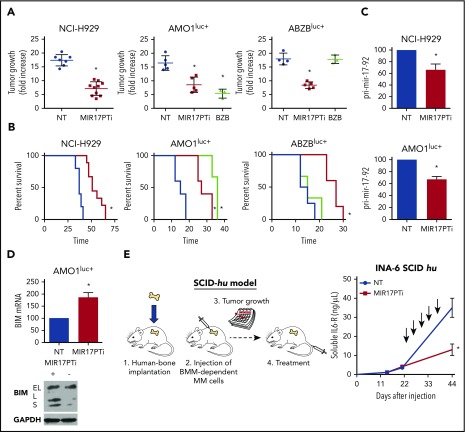
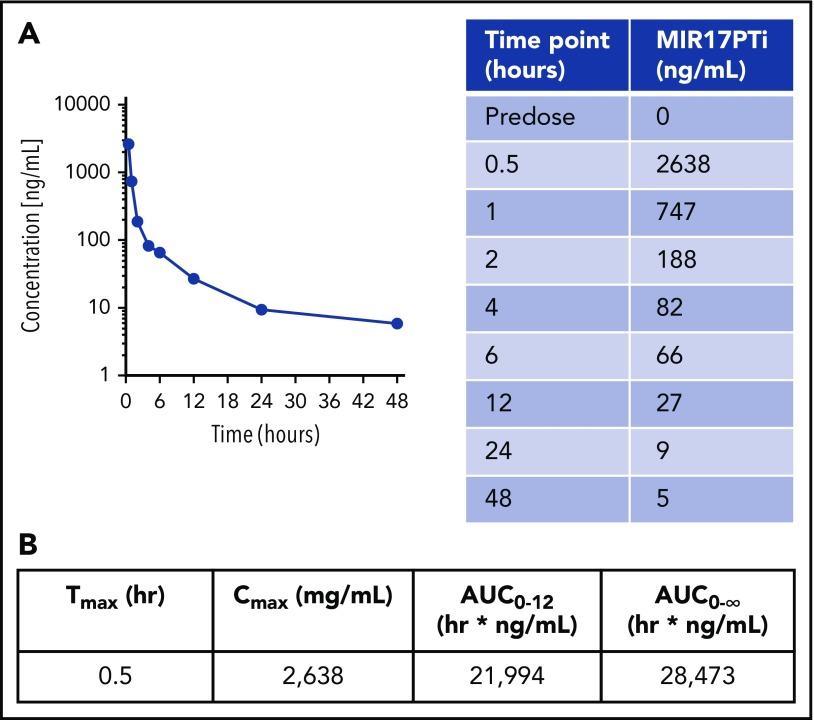
The microRNA (miRNA) cluster miR-17-92 is oncogenic and represents a valuable therapeutic target in c-MYC (MYC)-driven malignancies. Here, we developed novel LNA gapmeR antisense oligonucleotides (ASOs) to induce ribonuclease H-mediated degradation of MIR17HG primary transcripts and consequently prevent biogenesis of miR-17-92 miRNAs (miR-17-92s). The leading LNA ASO, MIR17PTi, impaired proliferation of several cancer cell lines (n = 48) established from both solid and hematologic tumors by on-target antisense activity, more effectively as compared with miR-17-92 inhibitors. By focusing on multiple myeloma (MM), we found that MIR17PTi triggers apoptosis via impairment of homeostatic MYC/miR-17-92 feed-forward loops (FFLs) in patient-derived MM cells and induces MYC-dependent synthetic lethality. We show that alteration of a BIM-centered FFL is instrumental for MIR17PTi to induce cytotoxicity in MM cells. MIR17PTi exerts strong in vivo antitumor activity in nonobese diabetic severe combined immunodeficient mice bearing clinically relevant models of MM, with advantageous safety and pharmacokinetic profiles in nonhuman primates. Altogether, MIR17PTi is a novel pharmacological tool to be tested in early-phase clinical trials against MM and other MYC-driven malignancies.
Conflict of interest statement
Conflict-of-interest disclosure: N.M.F. is an employee of Exiqon. The remaining authors declare no competing financial interests.
Figures
Comment in
-
A miRaculous new therapy in myeloma?Blood. 2018 Sep 6;132(10):983-985. doi: 10.1182/blood-2018-07-864207. Blood. 2018. PMID: 30190351 No abstract available.
References
-
- Ott G, Rosenwald A, Campo E. Understanding MYC-driven aggressive B-cell lymphomas: pathogenesis and classification. Blood. 2013;122(24):3884-3891. - PubMed
