

Lipid and Non-lipid Factors Affecting Macrophage Dysfunction and Inflammation in Atherosclerosis
- PMID: 29997514
- PMCID: PMC6029489
- DOI: 10.3389/fphys.2018.00654
Lipid and Non-lipid Factors Affecting Macrophage Dysfunction and Inflammation in Atherosclerosis
Abstract
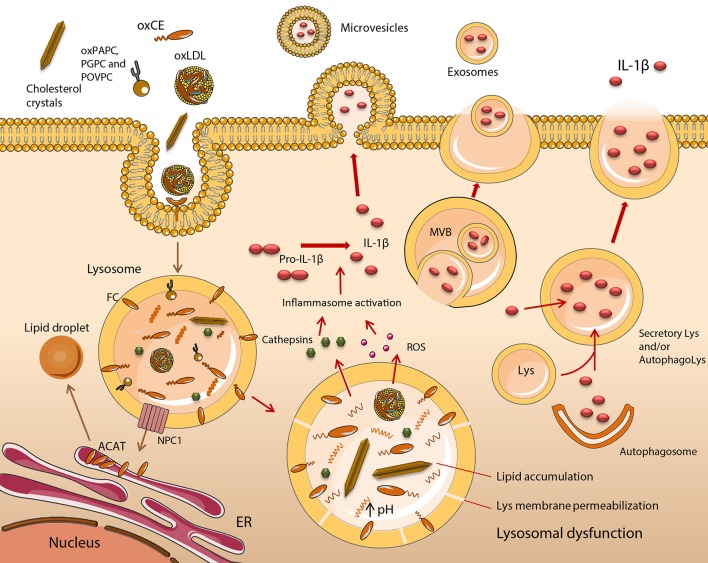
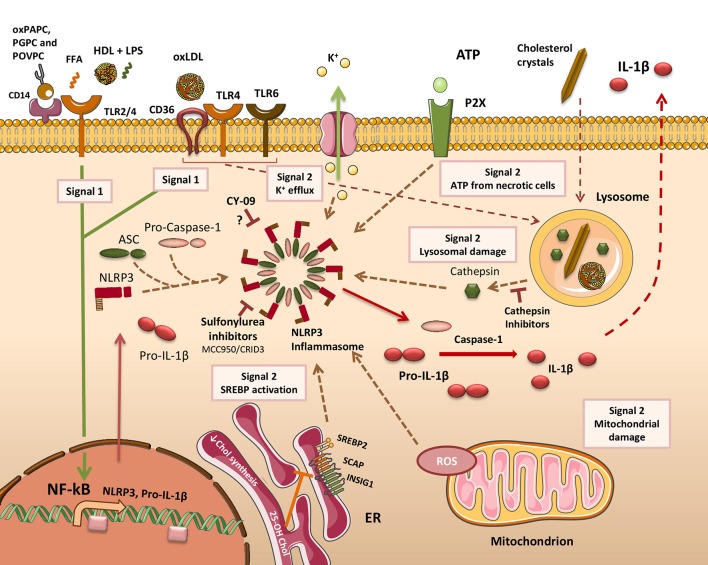
Atherosclerosis is a chronic inflammatory disease and a leading cause of human mortality. The lesional microenvironment contains a complex accumulation of variably oxidized lipids and cytokines. Infiltrating monocytes become polarized in response to these stimuli, resulting in a broad spectrum of macrophage phenotypes. The extent of lipid loading in macrophages influences their phenotype and consequently their inflammatory status. In response to excess atherogenic ligands, many normal cell processes become aberrant following a loss of homeostasis. This can have a direct impact upon the inflammatory response, and conversely inflammation can lead to cell dysfunction. Clear evidence for this exists in the lysosomes, endoplasmic reticulum and mitochondria of atherosclerotic macrophages, the principal lesional cell type. Furthermore, several intrinsic cell processes become dysregulated under lipidotic conditions. Therapeutic strategies aimed at restoring cell function under disease conditions are an ongoing coveted aim. Macrophages play a central role in promoting lesional inflammation, with plaque progression and stability being directly proportional to macrophage abundance. Understanding how mixtures or individual lipid species regulate macrophage biology is therefore a major area of atherosclerosis research. In this review, we will discuss how the myriad of lipid and lipoprotein classes and products used to model atherogenic, proinflammatory immune responses has facilitated a greater understanding of some of the intricacies of chronic inflammation and cell function. Despite this, lipid oxidation produces a complex mixture of products and with no single or standard method of derivatization, there exists some variation in the reported effects of certain oxidized lipids. Likewise, differences in the methods used to generate macrophages in vitro may also lead to variable responses when apparently identical lipid ligands are used. Consequently, the complexity of reported macrophage phenotypes has implications for our understanding of the metabolic pathways, processes and shifts underpinning their activation and inflammatory status. Using oxidized low density lipoproteins and its oxidized cholesteryl esters and phospholipid constituents to stimulate macrophage has been hugely valuable, however there is now an argument that only working with low complexity lipid species can deliver the most useful information to guide therapies aimed at controlling atherosclerosis and cardiovascular complications.
Keywords: atherosclerosis; chronic inflammation; lysosome dysfunction; macrophage heterogeneity; oxidized lipids.
Figures
References
-
- Bisgaard L. S., Mogensen C. K., Rosendahl A., Cucak H., Nielsen L. B., Rasmussen S. E., et al. (2016). Bone marrow-derived and peritoneal macrophages have different inflammatory response to oxLDL and M1/M2 marker expression - implications for atherosclerosis research. Sci. Rep. 6:35234. 10.1038/srep35234 - DOI - PMC - PubMed
-
- Black R. A., Kronheim S. R., Cantrell M., Deeley M. C., March C. J., Prickett K. S., et al. (1988). Generation of biologically active interleukin-1 beta by proteolytic cleavage of the inactive precursor. J. Biol. Chem., 263, 9437–9442. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources