

Valid molecular dynamics simulations of human hemoglobin require a surprisingly large box size
- PMID: 29998846
- PMCID: PMC6042964
- DOI: 10.7554/eLife.35560
Valid molecular dynamics simulations of human hemoglobin require a surprisingly large box size
Abstract
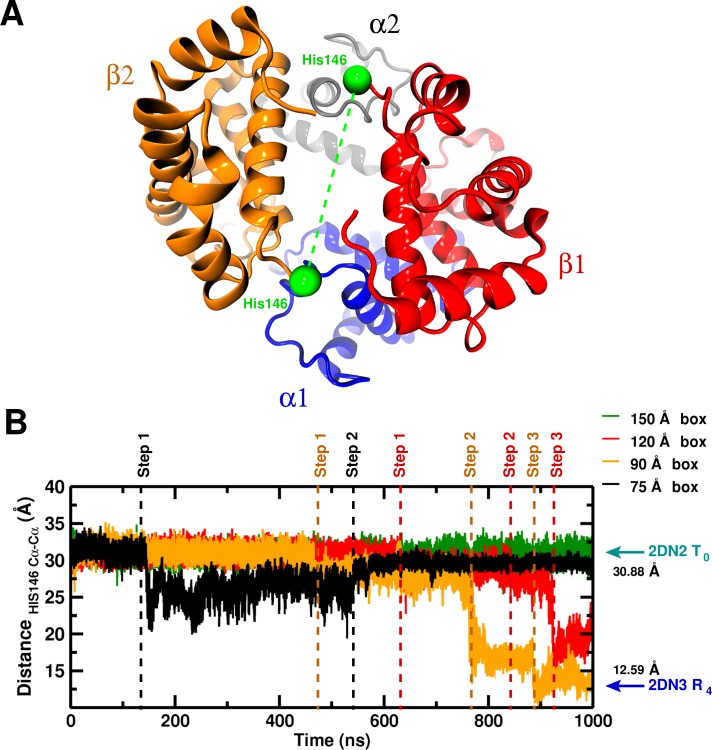
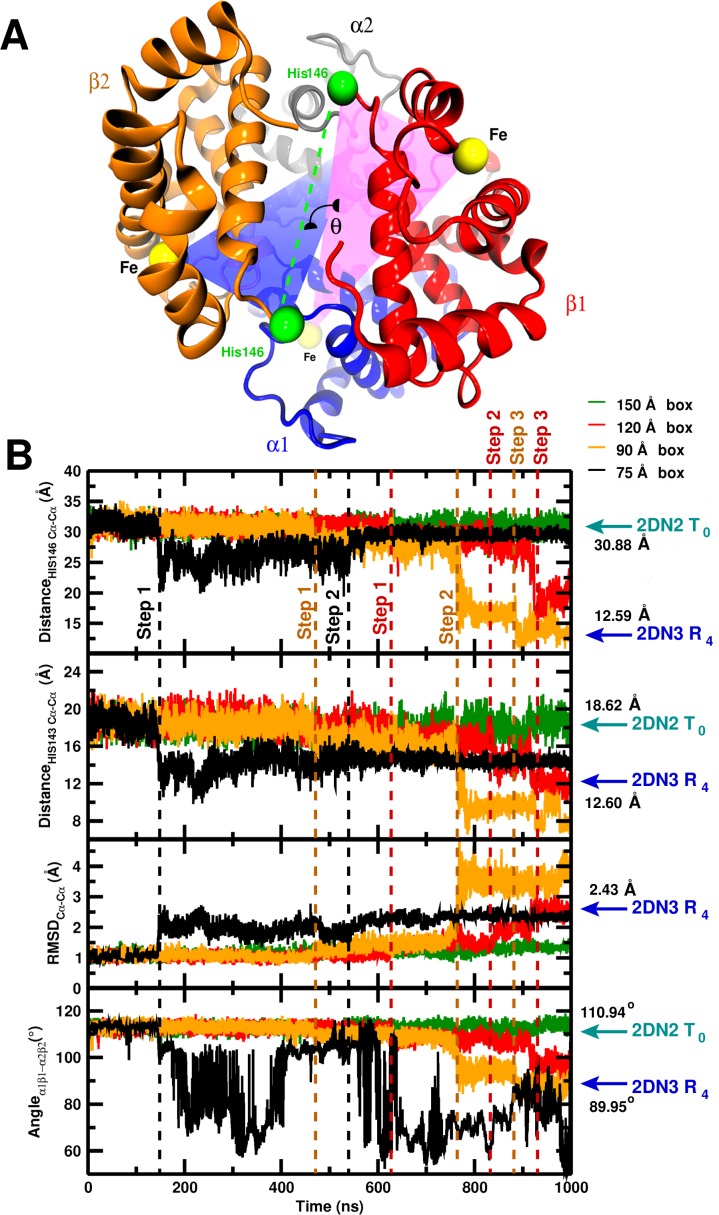
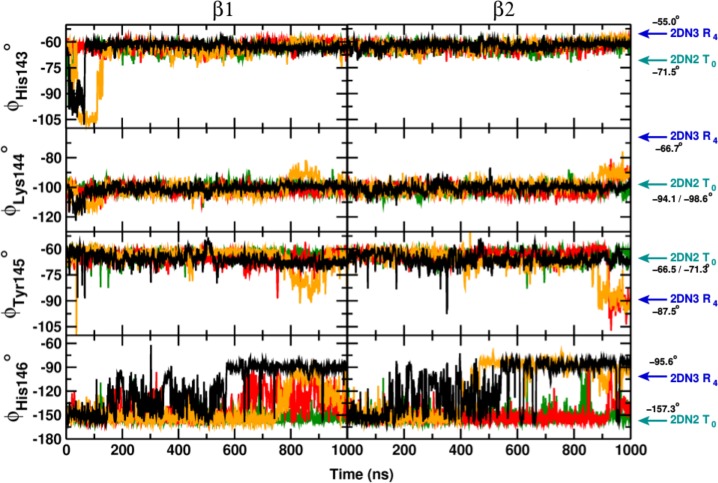
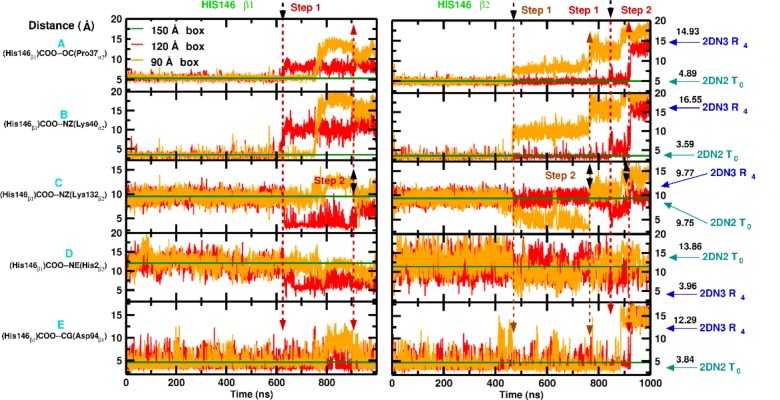
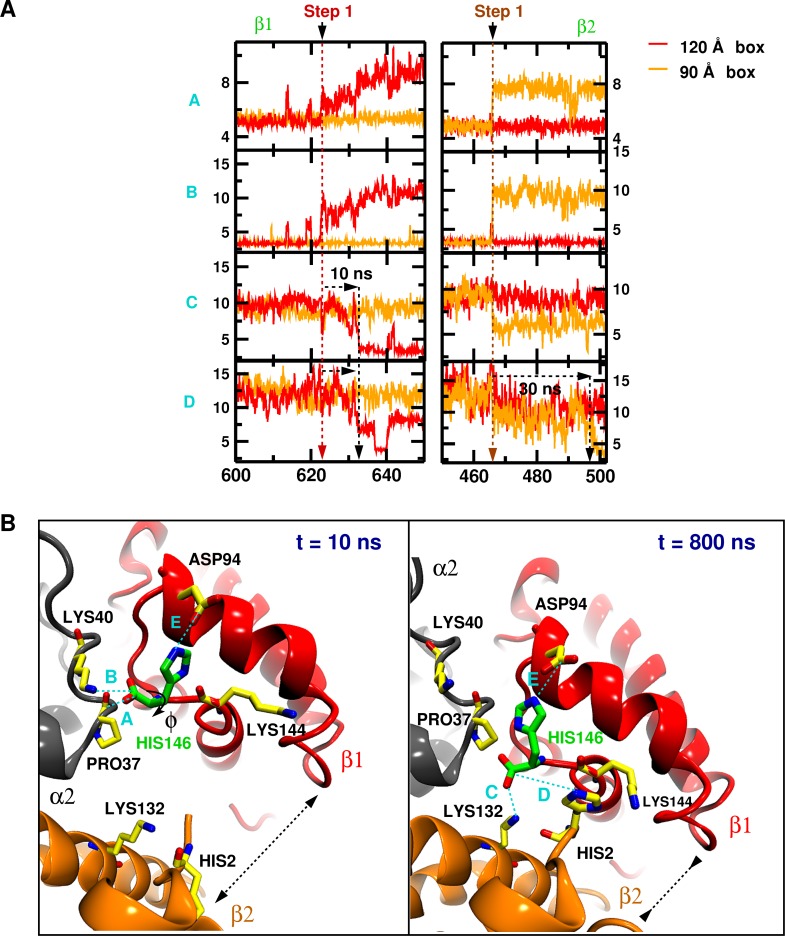
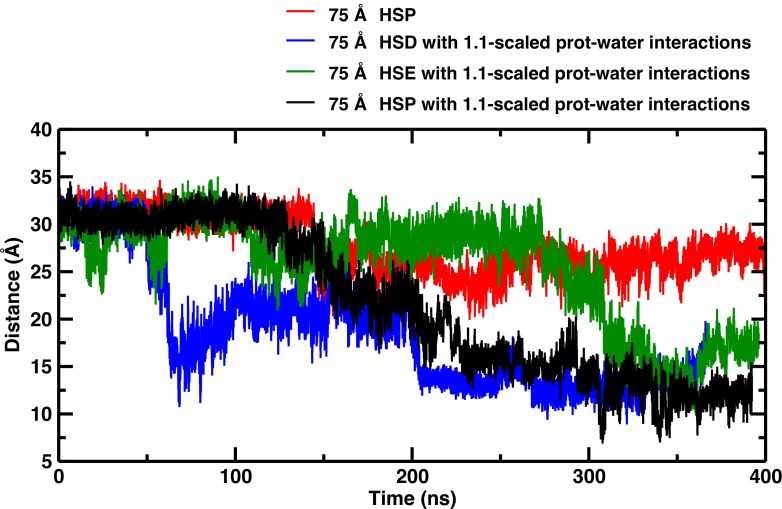
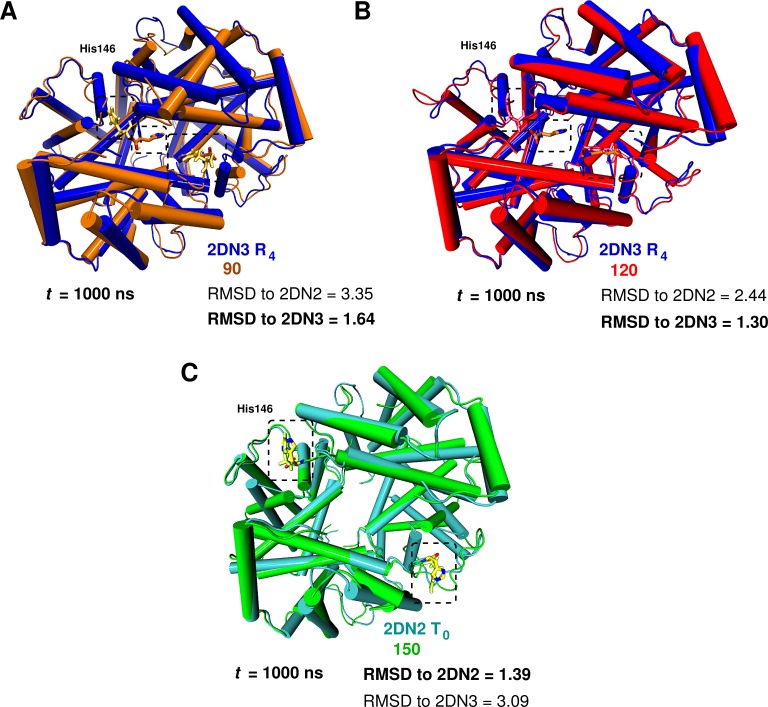
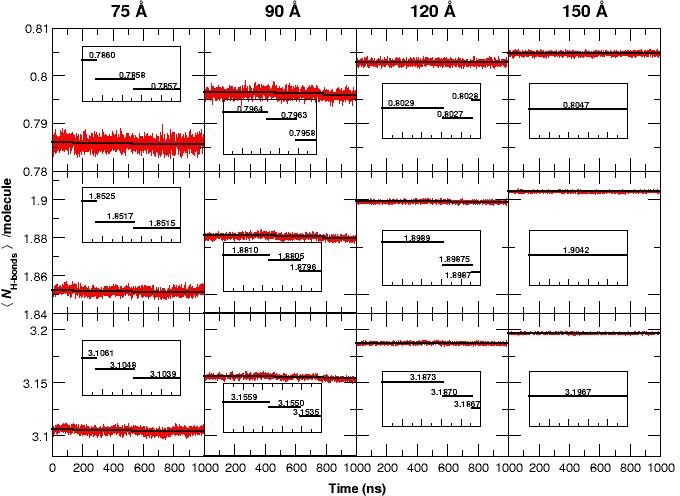
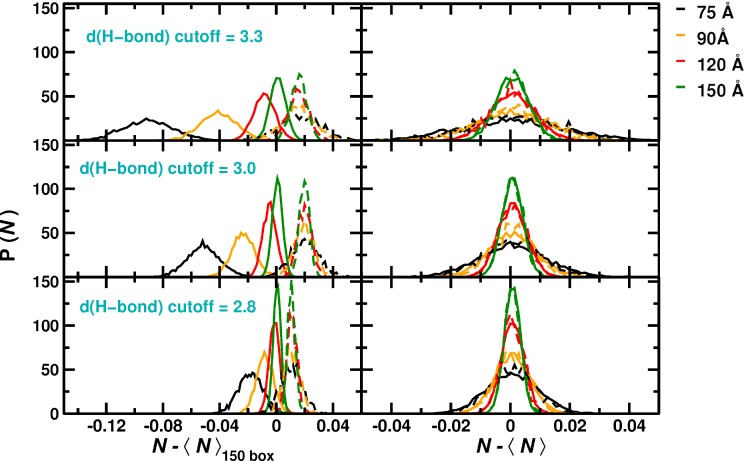
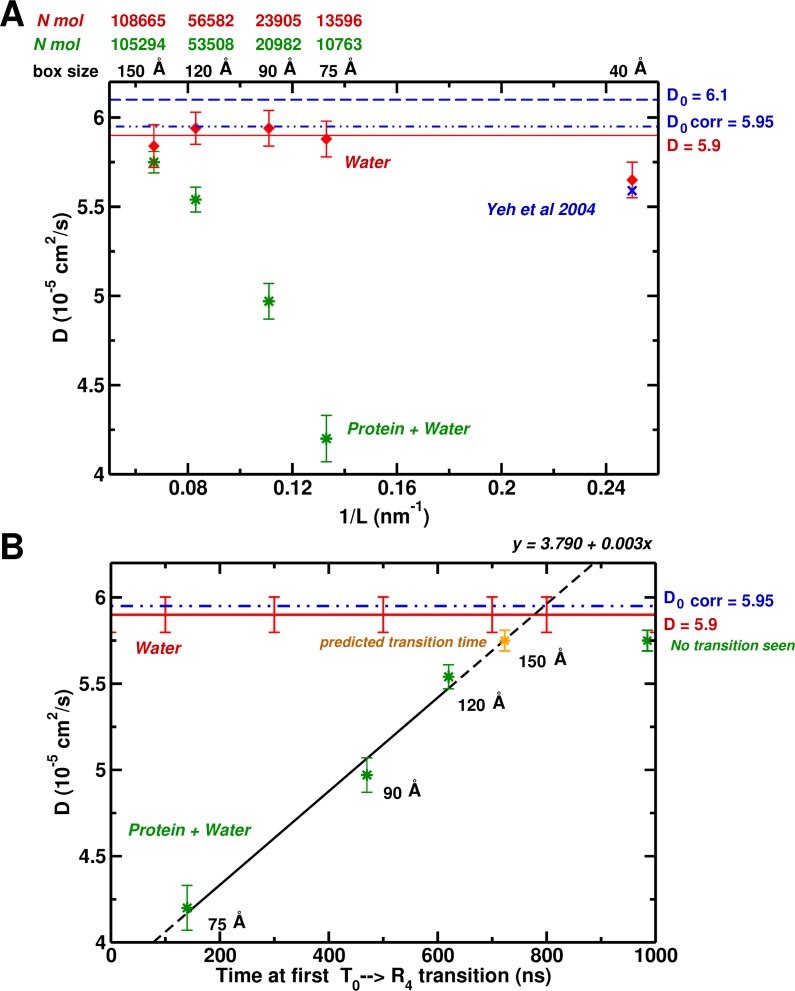
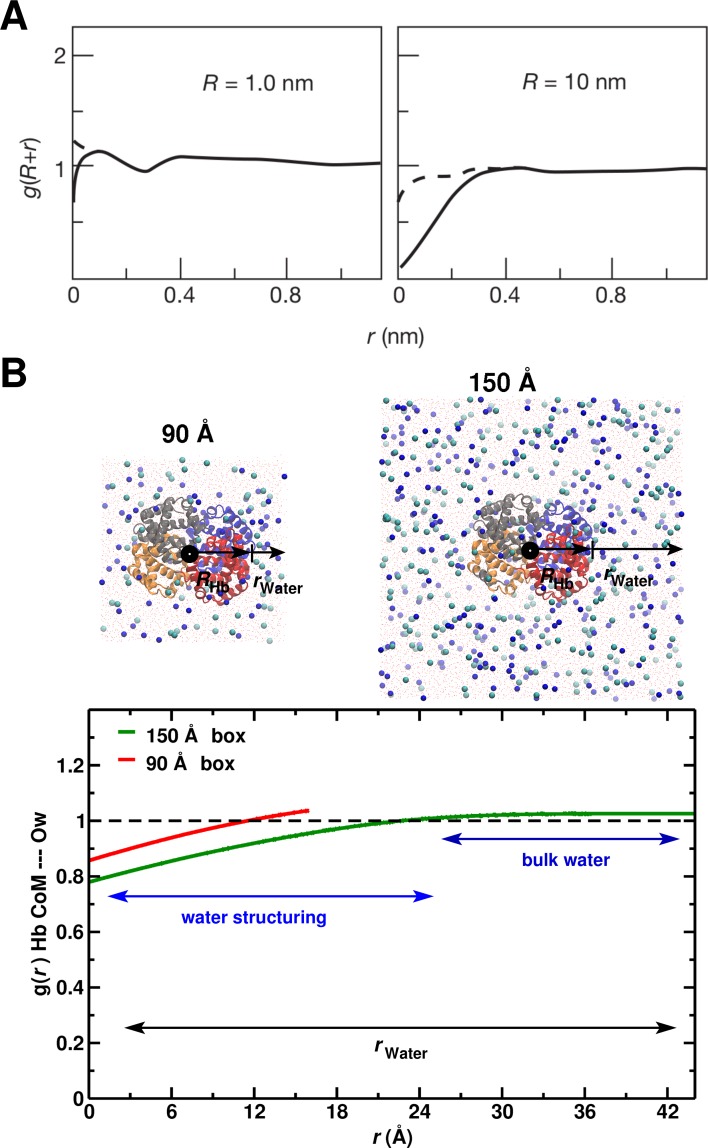
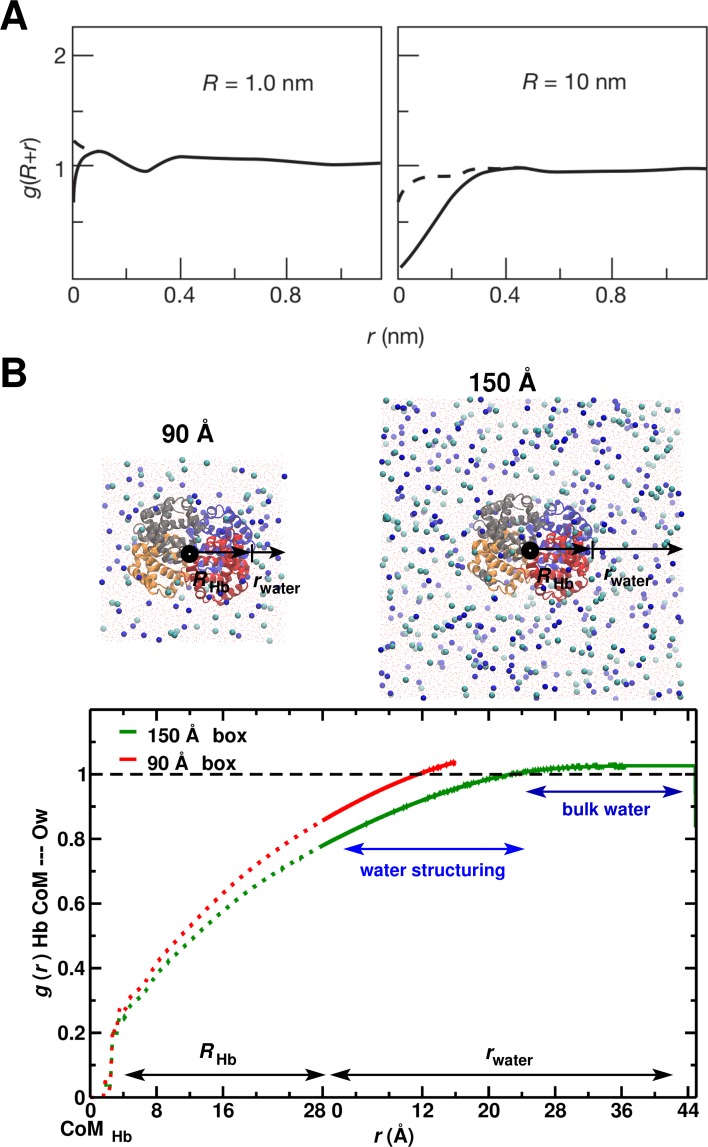
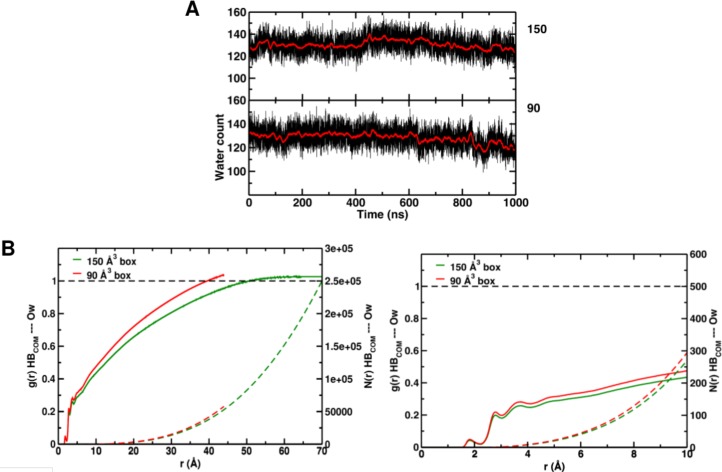
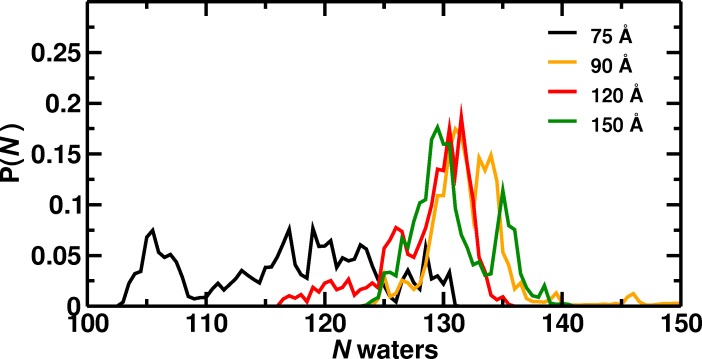
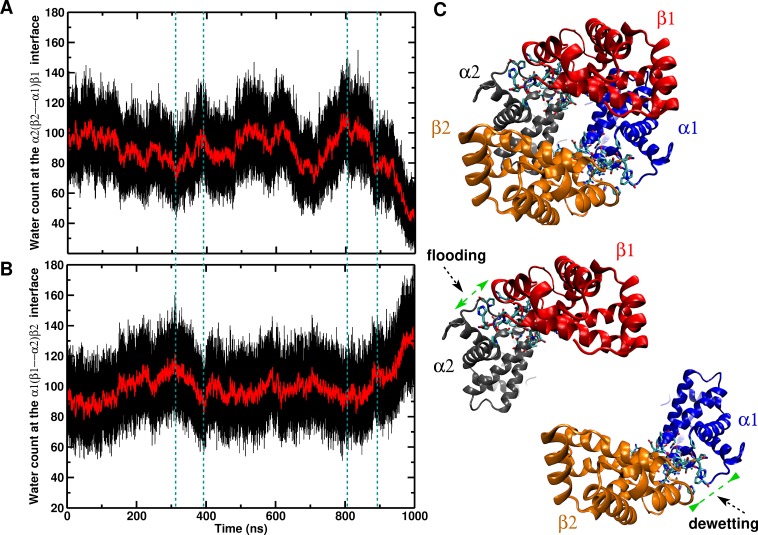
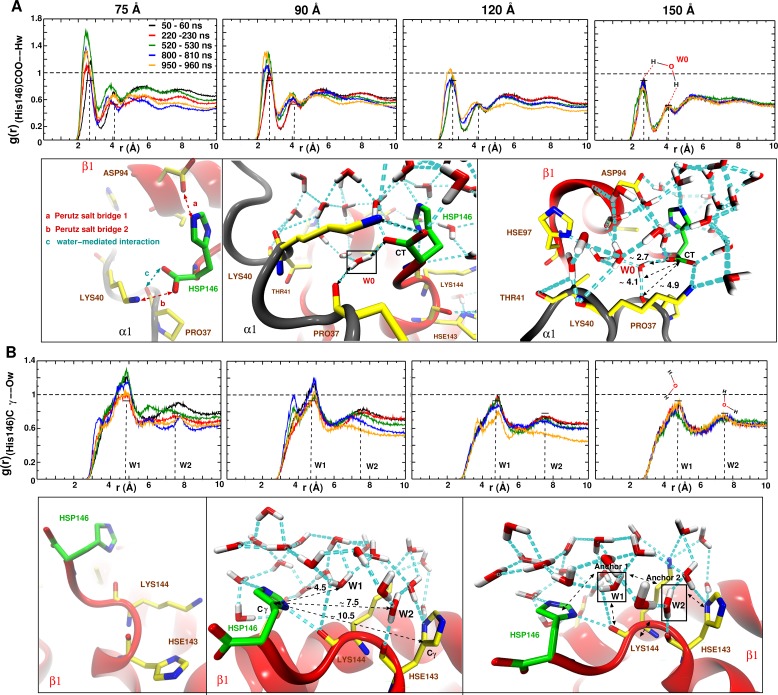
Recent molecular dynamics (MD) simulations of human hemoglobin (Hb) give results in disagreement with experiment. Although it is known that the unliganded (T[Formula: see text]) and liganded (R[Formula: see text]) tetramers are stable in solution, the published MD simulations of T[Formula: see text] undergo a rapid quaternary transition to an R-like structure. We show that T[Formula: see text] is stable only when the periodic solvent box contains ten times more water molecules than the standard size for such simulations. The results suggest that such a large box is required for the hydrophobic effect, which stabilizes the T[Formula: see text] tetramer, to be manifested. Even in the largest box, T[Formula: see text] is not stable unless His146 is protonated, providing an atomistic validation of the Perutz model. The possibility that extra large boxes are required to obtain meaningful results will have to be considered in evaluating existing and future simulations of a wide range of systems.
Keywords: diffusion constant; hemoglobin; hydrophobic effect; molecular biophysics; molecular dynamics; none; simulation box size; structural biology.
© 2018, El Hage et al.
Conflict of interest statement
KE, FH, PG, MM, MK No competing interests declared
Figures
Comment in
-
Comment on 'Valid molecular dynamics simulations of human hemoglobin require a surprisingly large box size'.Elife. 2019 Jun 20;8:e44718. doi: 10.7554/eLife.44718. Elife. 2019. PMID: 31219782 Free PMC article.
-
Response to comment on 'Valid molecular dynamics simulations of human hemoglobin require a surprisingly large box size'.Elife. 2019 Jun 20;8:e45318. doi: 10.7554/eLife.45318. Elife. 2019. PMID: 31219783 Free PMC article.
References
-
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. doi: 10.1016/j.softx.2015.06.001. - DOI
-
- Allen MP, Tildesley DJ. Computer Simulation of Liquids. Oxford, U.K: Clarendon Press; 1987.
-
- Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, Mackerell AD. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. Journal of Chemical Theory and Computation. 2012;8:3257–3273. doi: 10.1021/ct300400x. - DOI - PMC - PubMed
