

Alternative empirical Bayes models for adjusting for batch effects in genomic studies
- PMID: 30001694
- PMCID: PMC6044013
- DOI: 10.1186/s12859-018-2263-6
Alternative empirical Bayes models for adjusting for batch effects in genomic studies
Abstract
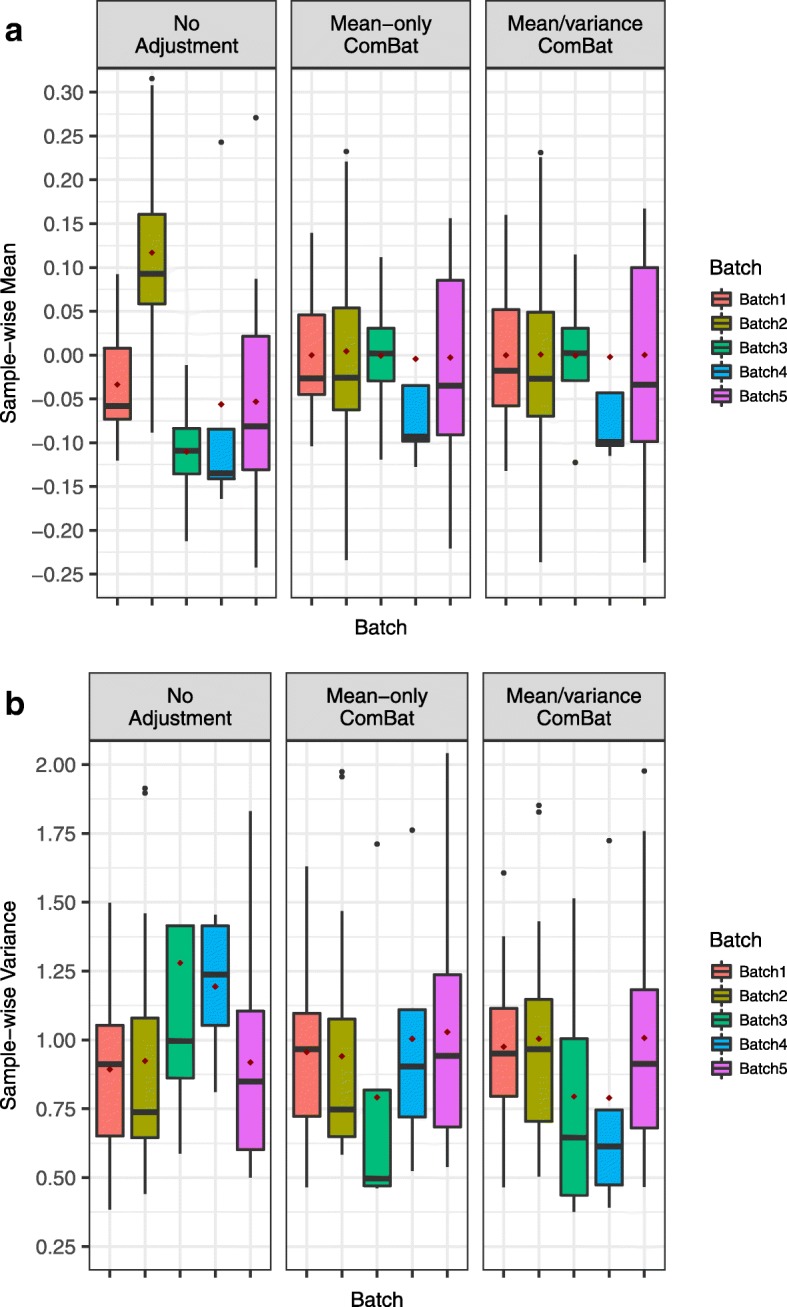
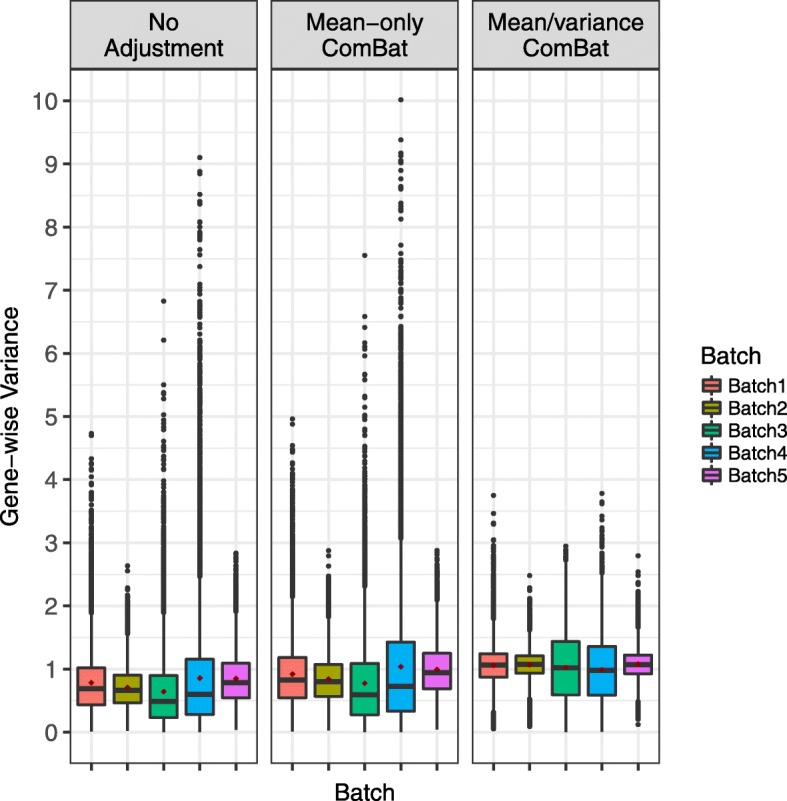
Background: Combining genomic data sets from multiple studies is advantageous to increase statistical power in studies where logistical considerations restrict sample size or require the sequential generation of data. However, significant technical heterogeneity is commonly observed across multiple batches of data that are generated from different processing or reagent batches, experimenters, protocols, or profiling platforms. These so-called batch effects often confound true biological relationships in the data, reducing the power benefits of combining multiple batches, and may even lead to spurious results in some combined studies. Therefore there is significant need for effective methods and software tools that account for batch effects in high-throughput genomic studies.
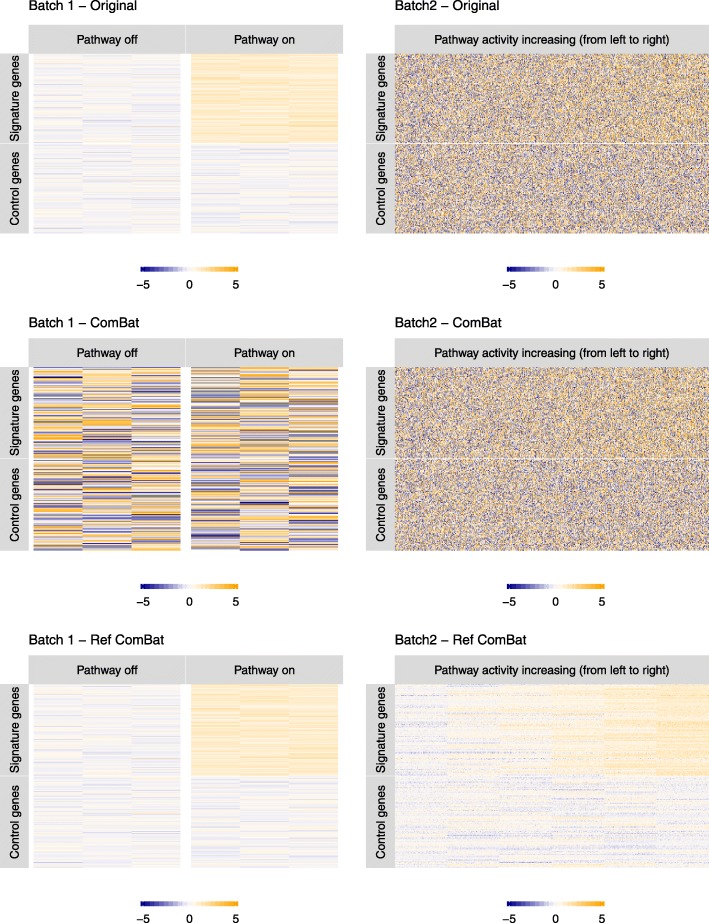
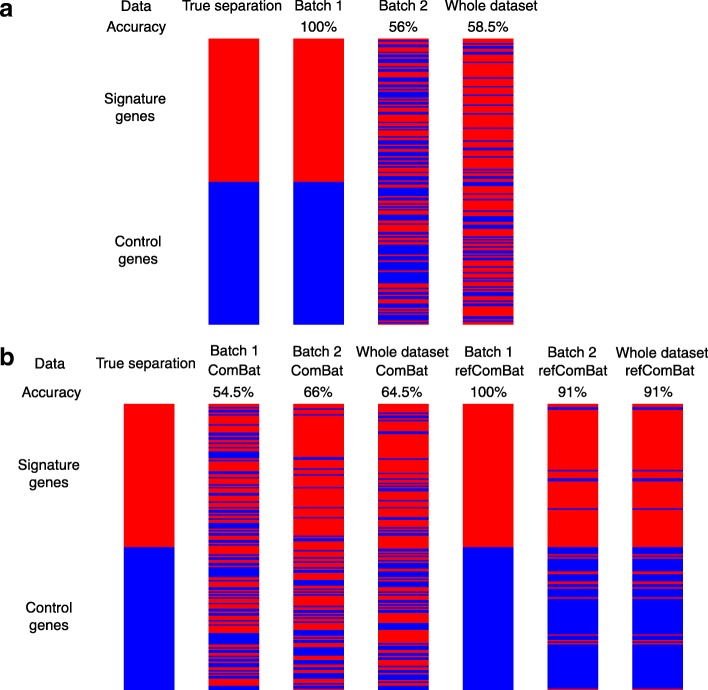
Results: Here we contribute multiple methods and software tools for improved combination and analysis of data from multiple batches. In particular, we provide batch effect solutions for cases where the severity of the batch effects is not extreme, and for cases where one high-quality batch can serve as a reference, such as the training set in a biomarker study. We illustrate our approaches and software in both simulated and real data scenarios.
Conclusions: We demonstrate the value of these new contributions compared to currently established approaches in the specified batch correction situations.
Keywords: Batch effects; Biomarker development; Data integration; Empirical Bayes models.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
