

The value of universally available raw NMR data for transparency, reproducibility, and integrity in natural product research
- PMID: 30003207
- PMCID: PMC6350634
- DOI: 10.1039/c7np00064b
The value of universally available raw NMR data for transparency, reproducibility, and integrity in natural product research
Erratum in
-
Correction: The value of universally available raw NMR data for transparency, reproducibility, and integrity in natural product research.Nat Prod Rep. 2019 Jan 1;36(1):248-249. doi: 10.1039/c8np90041h. Epub 2018 Nov 23. Nat Prod Rep. 2019. PMID: 30468235 Free PMC article.
Abstract
Covering: up to 2018With contributions from the global natural product (NP) research community, and continuing the Raw Data Initiative, this review collects a comprehensive demonstration of the immense scientific value of disseminating raw nuclear magnetic resonance (NMR) data, independently of, and in parallel with, classical publishing outlets. A comprehensive compilation of historic to present-day cases as well as contemporary and future applications show that addressing the urgent need for a repository of publicly accessible raw NMR data has the potential to transform natural products (NPs) and associated fields of chemical and biomedical research. The call for advancing open sharing mechanisms for raw data is intended to enhance the transparency of experimental protocols, augment the reproducibility of reported outcomes, including biological studies, become a regular component of responsible research, and thereby enrich the integrity of NP research and related fields.
Figures
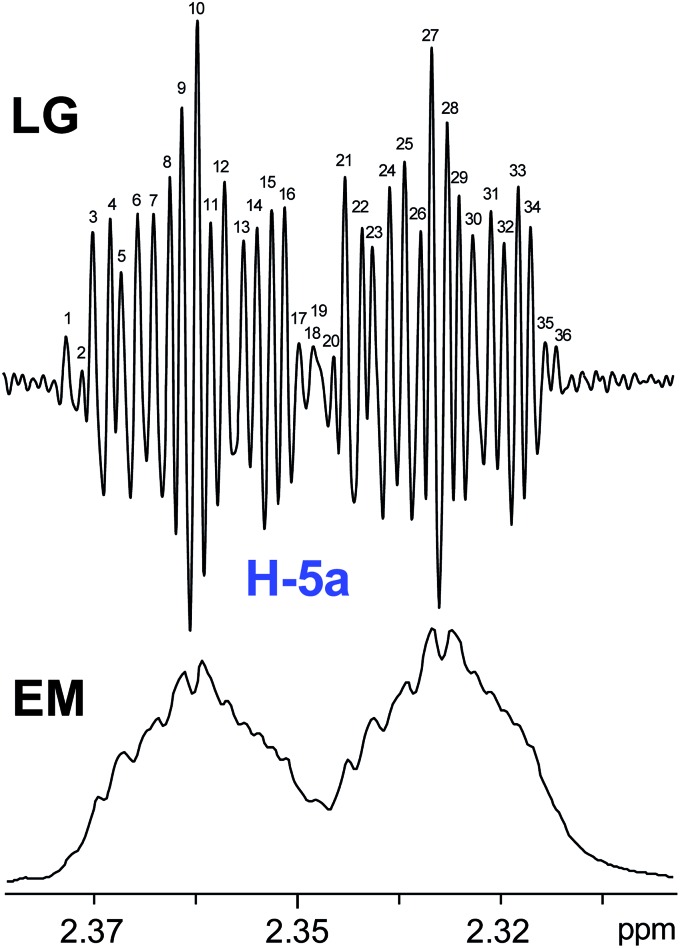
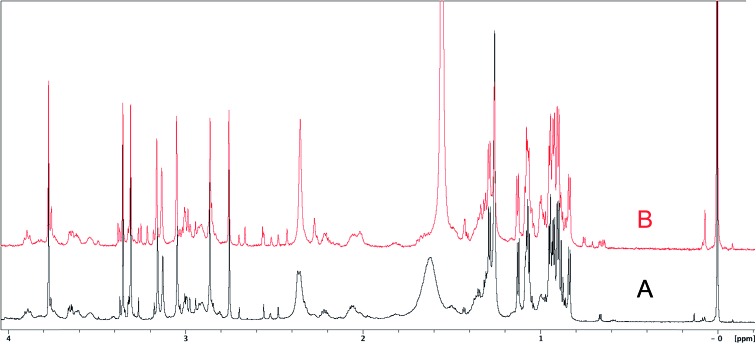
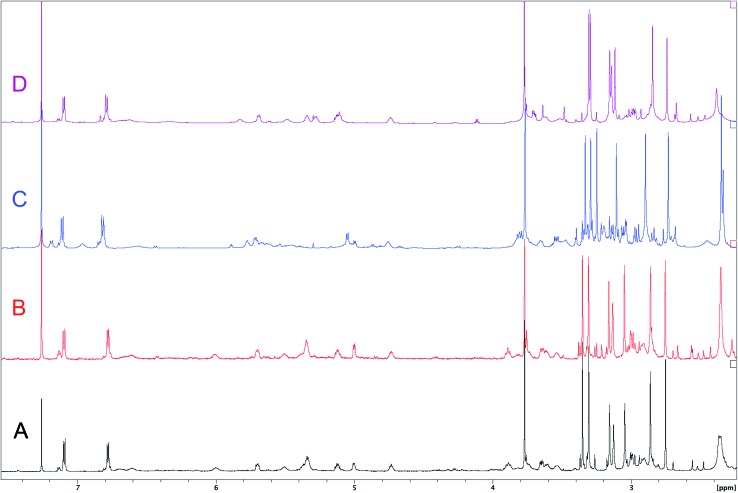
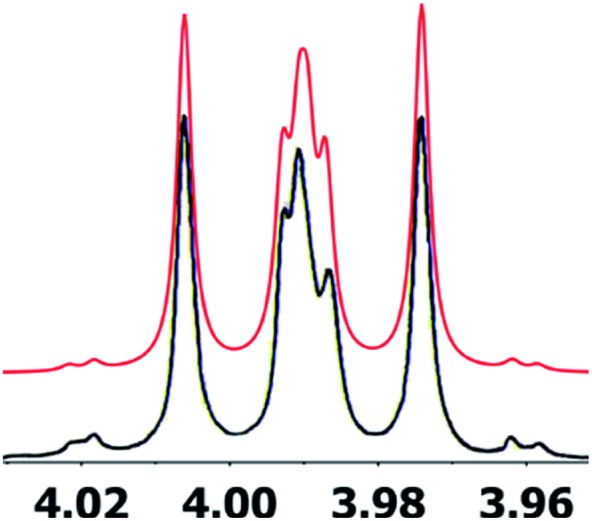

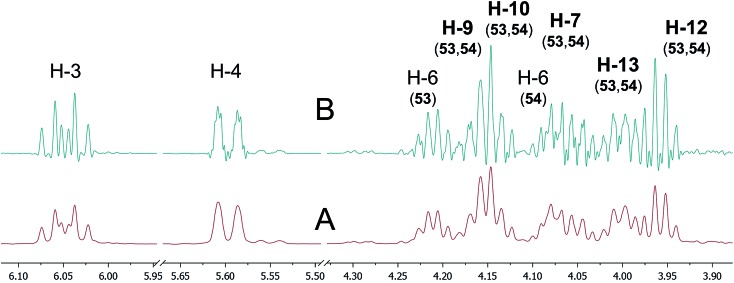
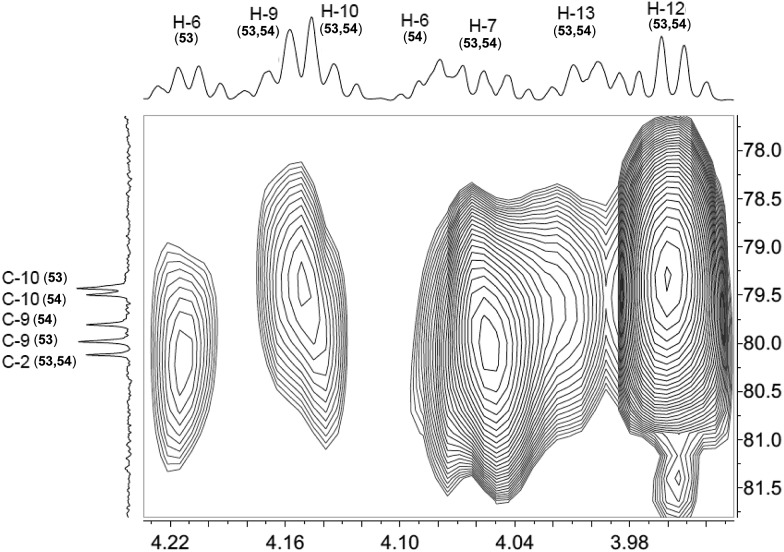
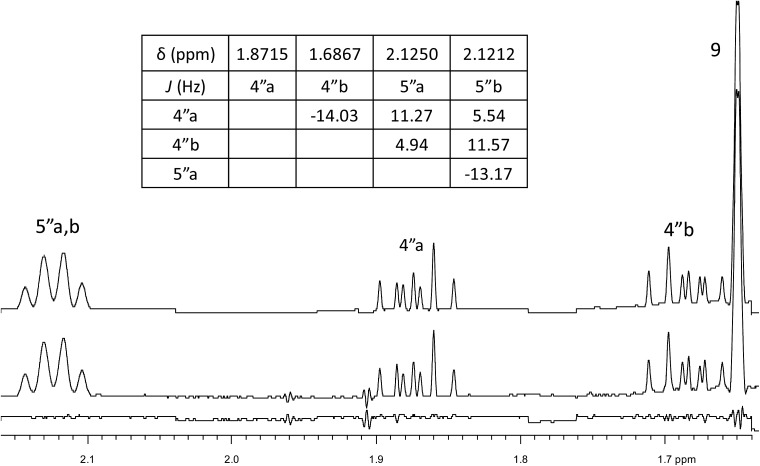
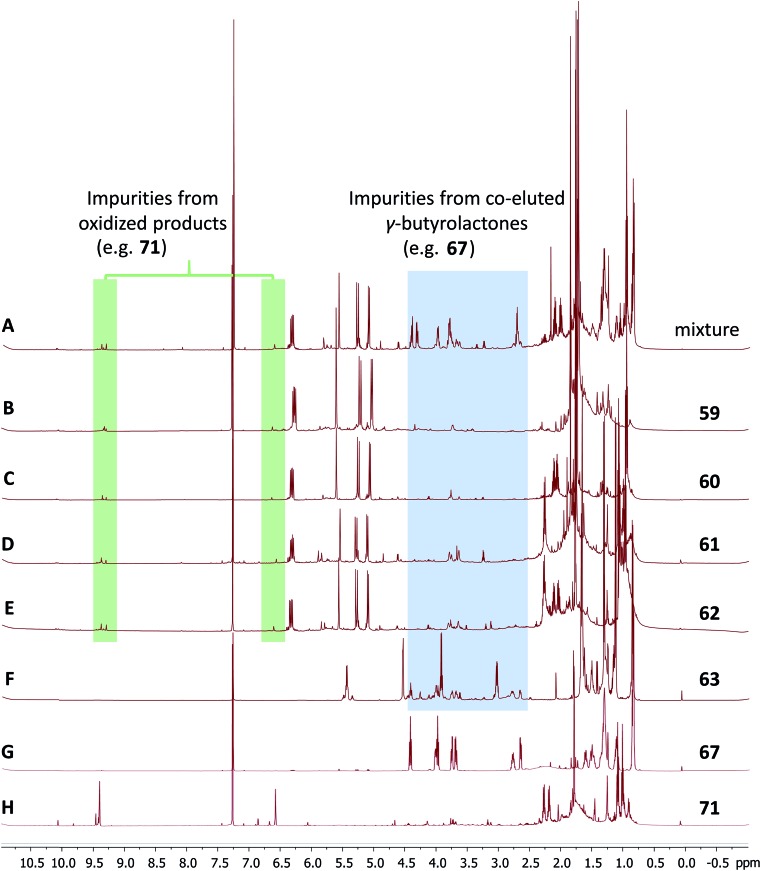
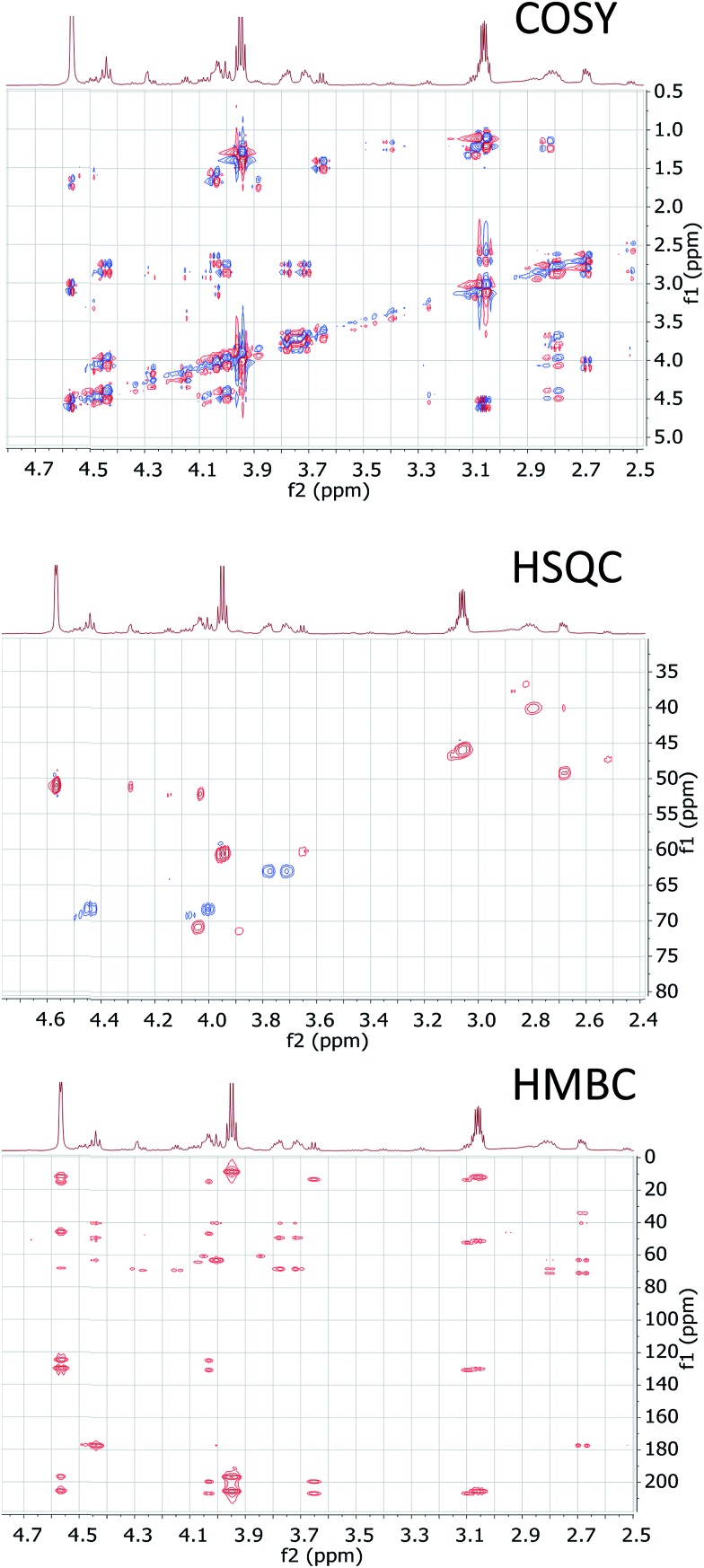
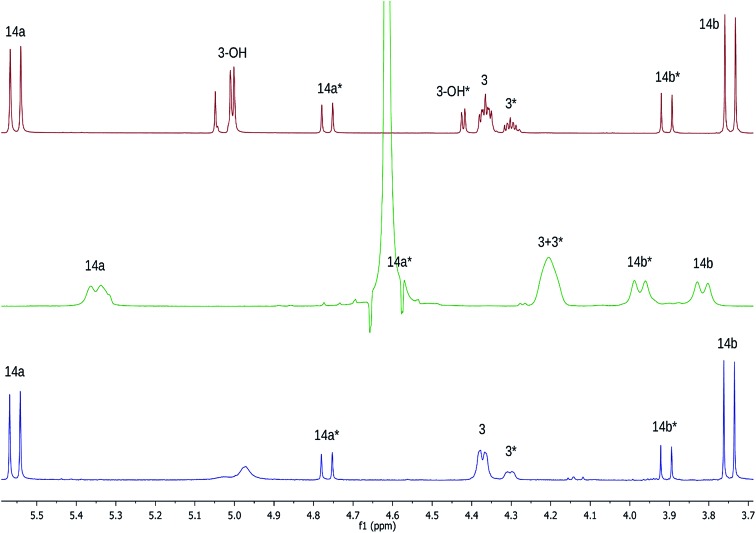
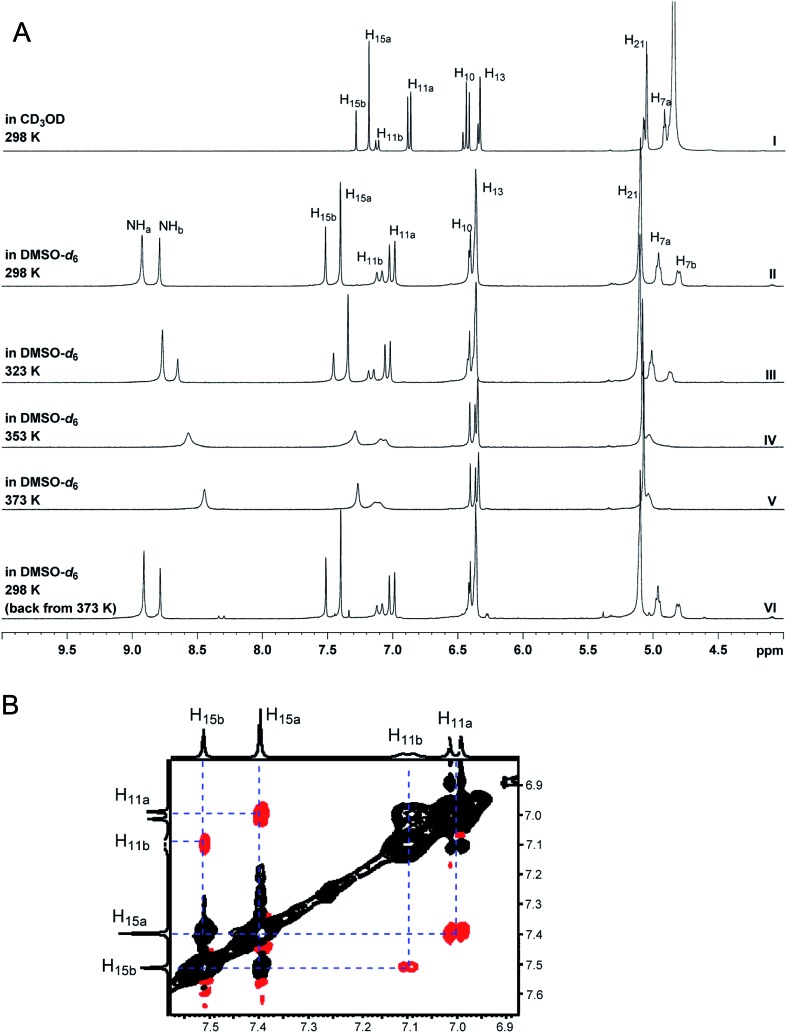

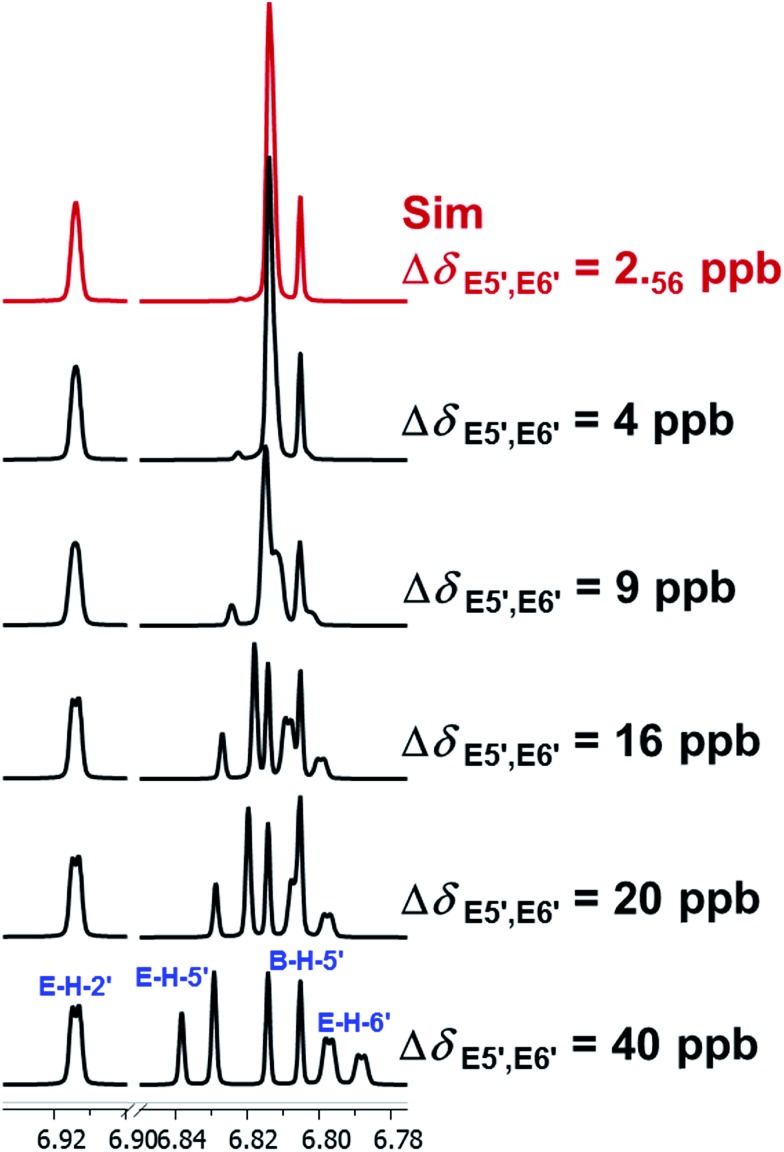
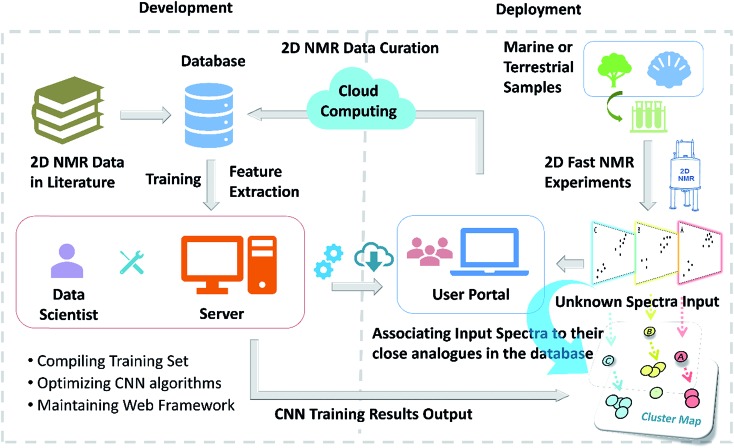
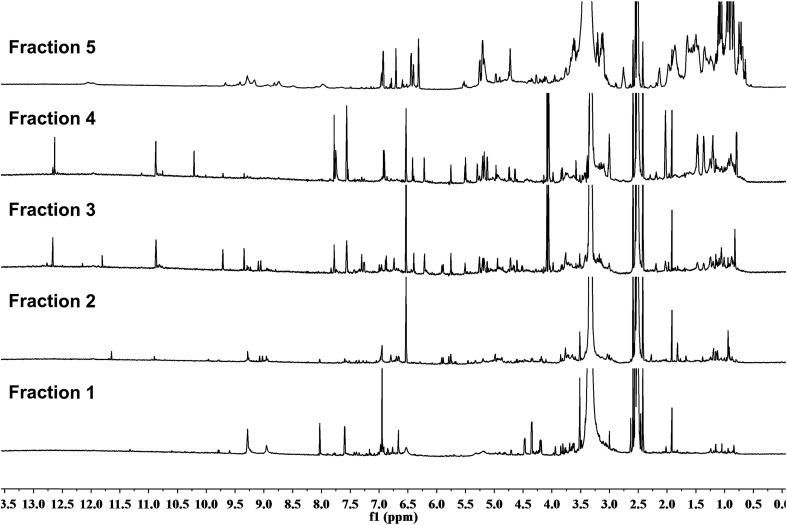
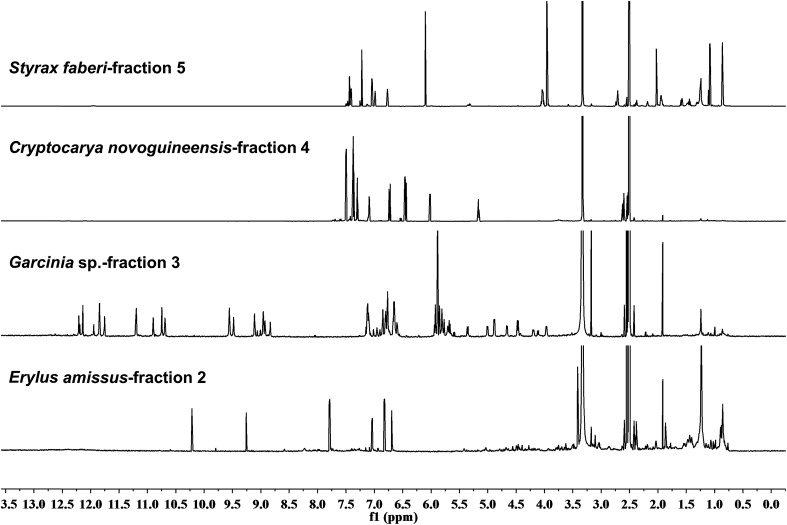
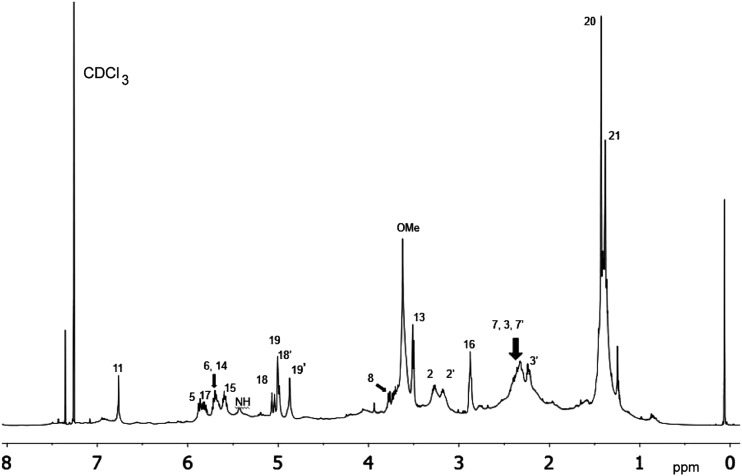
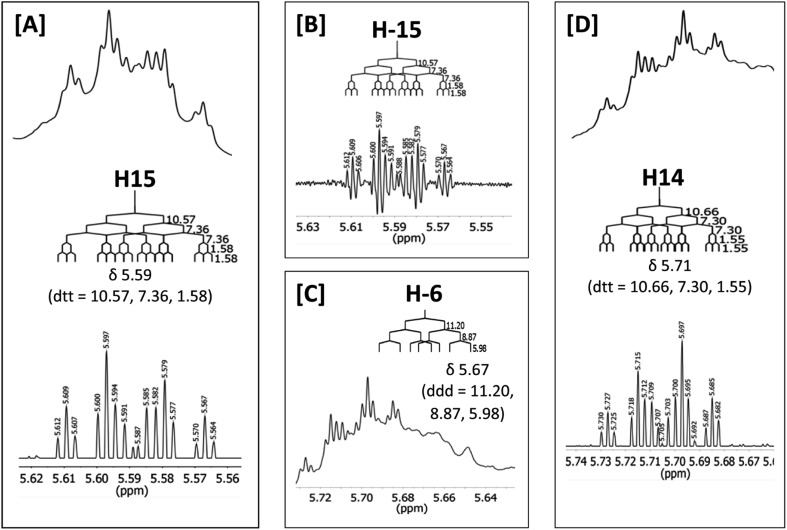
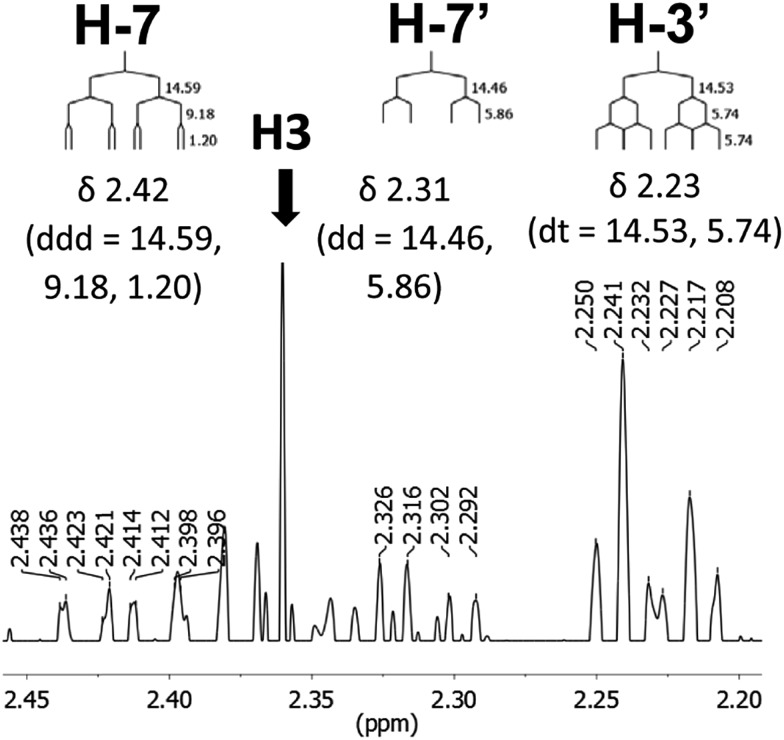
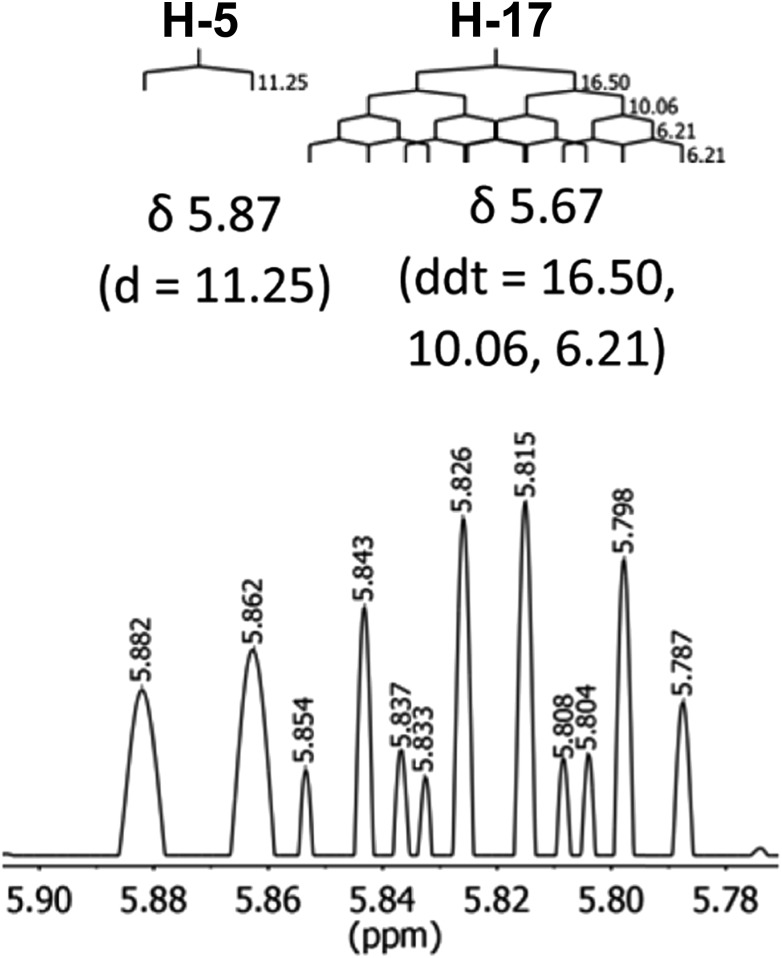

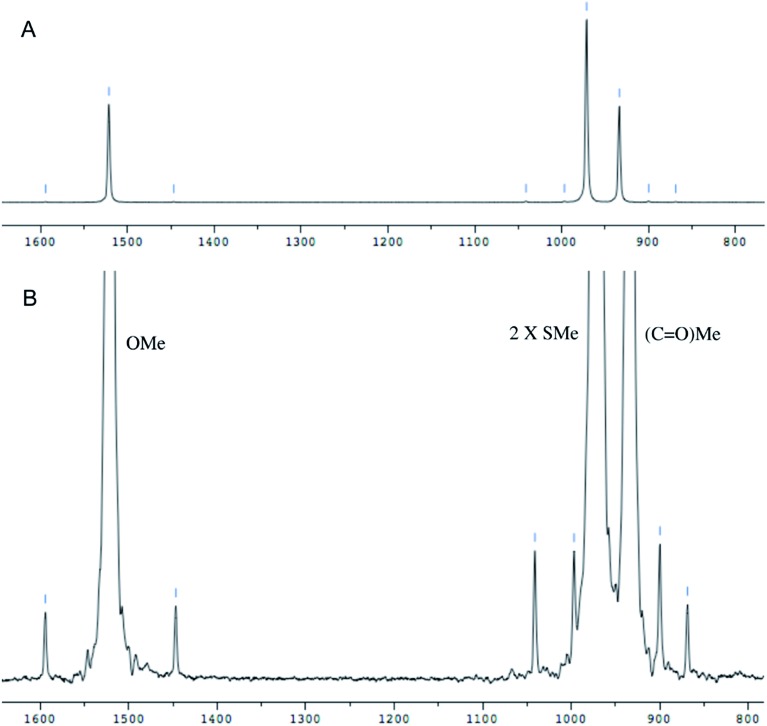


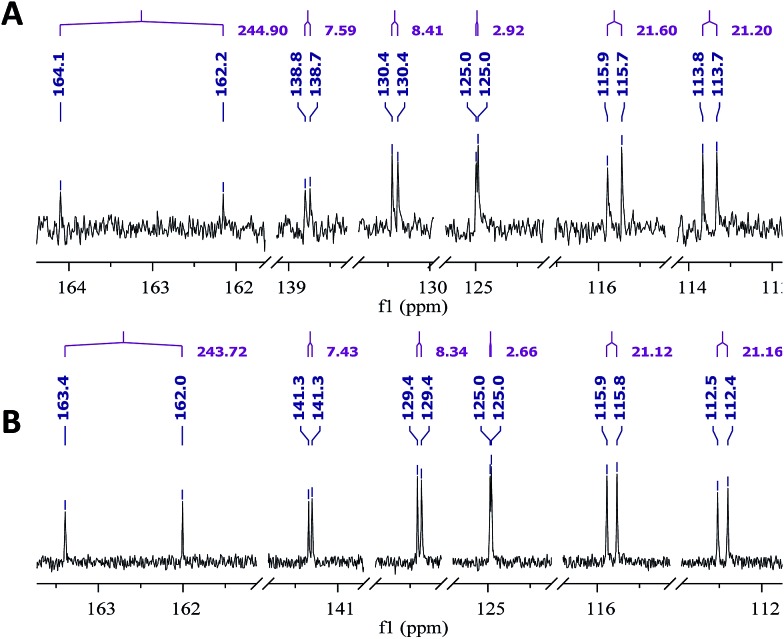
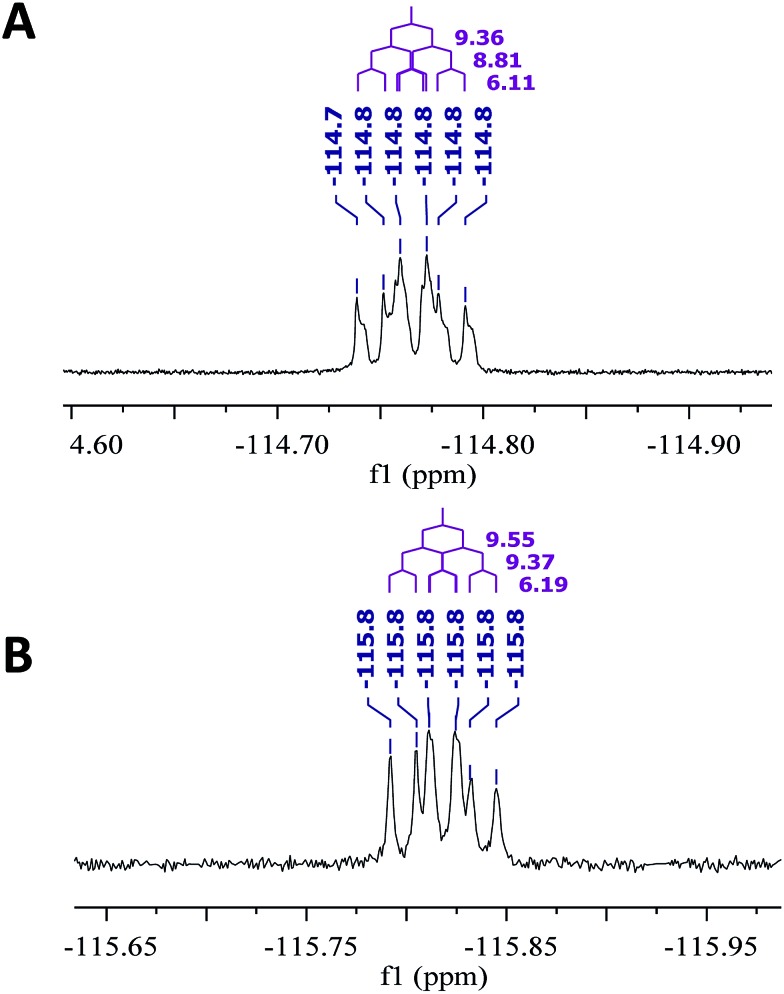




References
-
- Elyashberg M., Williams A. J. and Blinov K., Contemporary Computer-Assisted Approaches to Molecular Structure Elucidation, The Royal Society of Chemistry, Cambridge, 2012.
-
- Buevich A. V., Elyashberg M. E. J. Nat. Prod. 2016;79:3105–3116. - PubMed
-
- Lodewyk M. W., Soldi C., Jones P. B., Olmstead M. M., Rita J., Shaw J. T., Tantillo D. J. J. Am. Chem. Soc. 2012;134:18550–18553. - PubMed
-
- Kuhn S., Schlörer N. E. Magn. Reson. Chem. 2015;53:582–589. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous