

Single-Cell Multi-omics: An Engine for New Quantitative Models of Gene Regulation
- PMID: 30007833
- PMCID: PMC6097890
- DOI: 10.1016/j.tig.2018.06.001
Single-Cell Multi-omics: An Engine for New Quantitative Models of Gene Regulation
Abstract
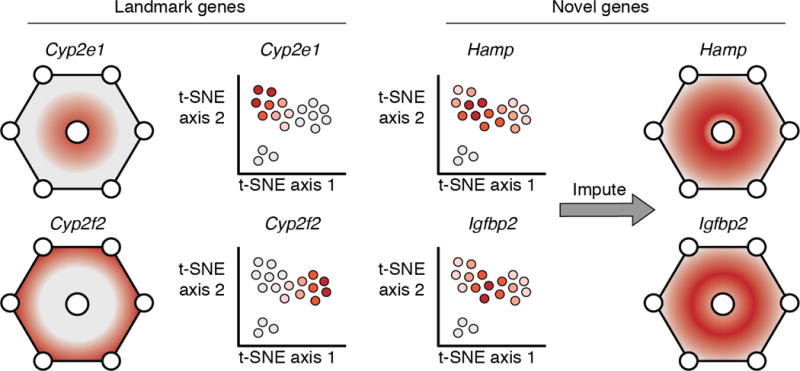
Cells in a multicellular organism fulfill specific functions by enacting cell-type-specific programs of gene regulation. Single-cell RNA sequencing technologies have provided a transformative view of cell-type-specific gene expression, the output of cell-type-specific gene regulatory programs. This review discusses new single-cell genomic technologies that complement single-cell RNA sequencing by providing additional readouts of cellular state beyond the transcriptome. We highlight regression models as a simple yet powerful approach to relate gene expression to other aspects of cellular state, and in doing so, gain insights into the biochemical mechanisms that are necessary to produce a given gene expression output.
Copyright © 2018 Elsevier Ltd. All rights reserved.
Figures


References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
