

Stress and aging act through common mechanisms to elicit neuroinflammatory priming
- PMID: 30009999
- PMCID: PMC6129421
- DOI: 10.1016/j.bbi.2018.07.012
Stress and aging act through common mechanisms to elicit neuroinflammatory priming
Abstract
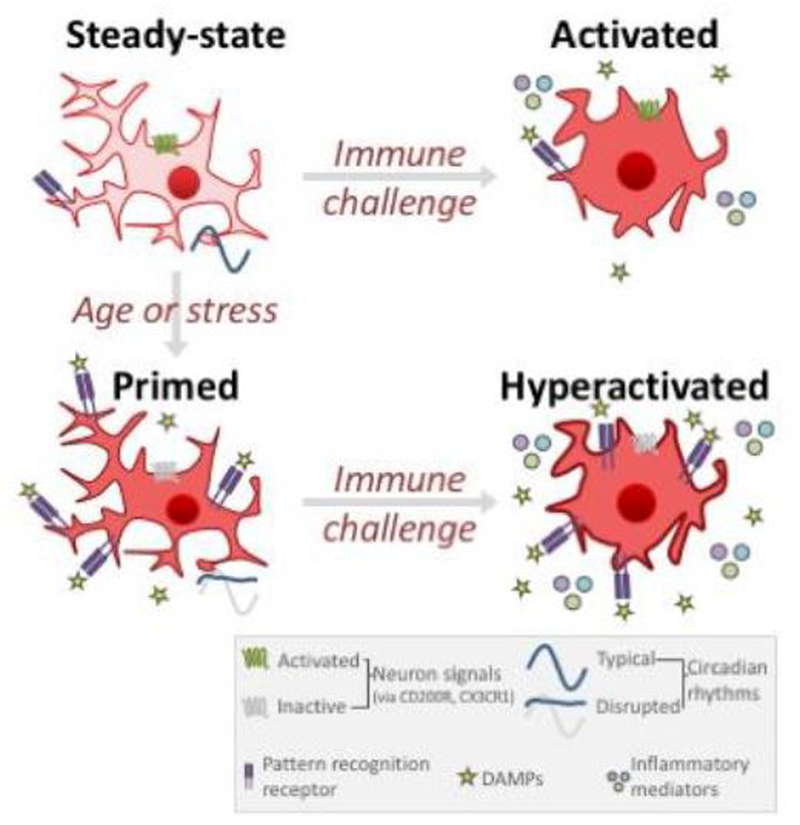
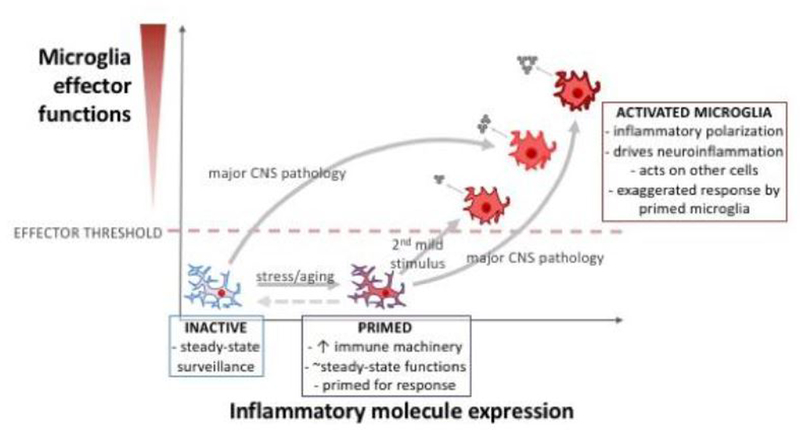
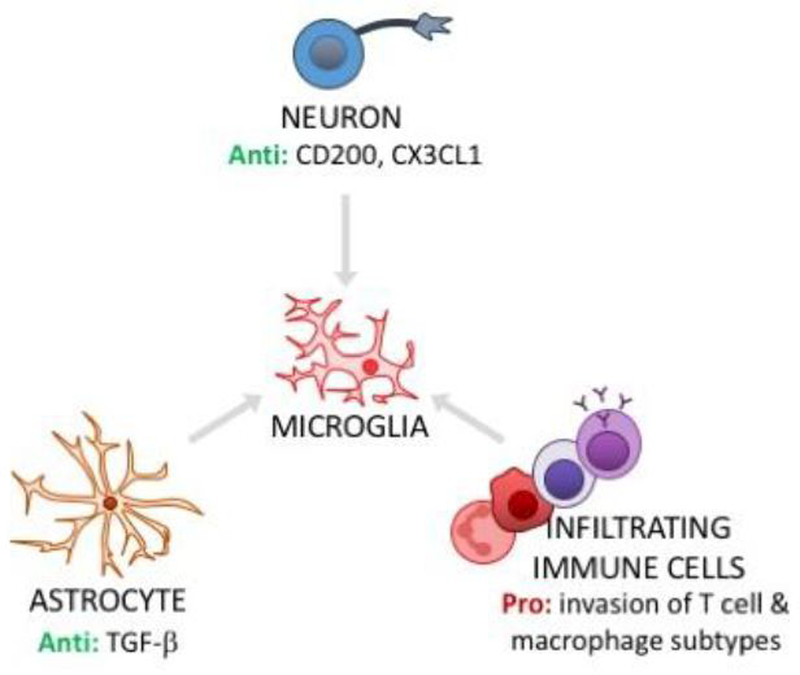
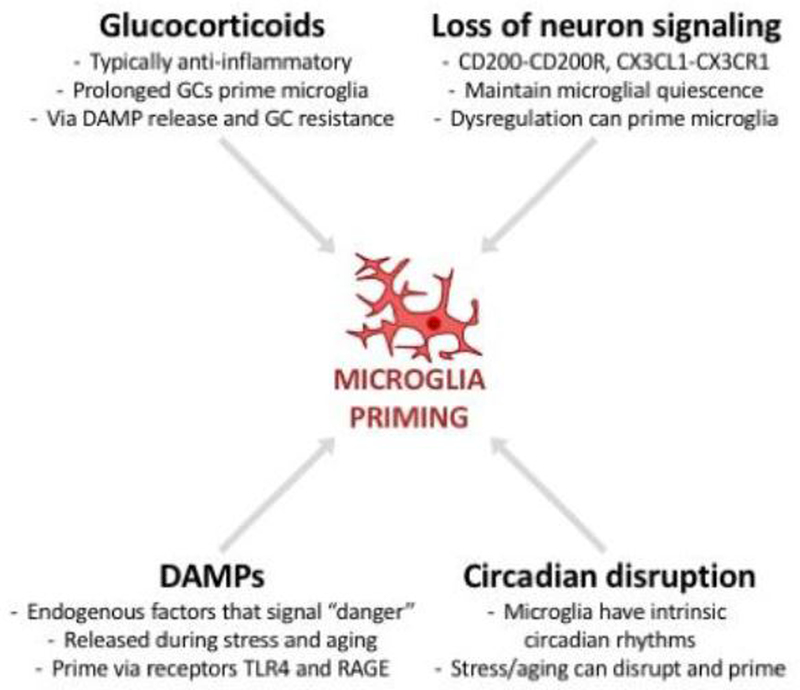
Over the course of an animal's lifespan, there is a protracted breakdown in basic homeostatic functions. Stressors (both psychological and physiological) can accelerate this process and compromise multiple homeostatic mechanisms. For example, both stress and aging can modulate neuroinflammatory function and cause a primed phenotype resulting in a heightened neuroinflammatory profile upon immune activation. Microglia, the brain's resident myeloid cell, produce "silent" immune machinery in response to stress and aging that does not cause immediate immune activation; rather, these changes prime the cell for a subsequent immune insult. Primed microglia exhibit a hyperinflammatory response upon immune activation that can exacerbate pathology. In this review, we will explore parallels between stress- and aging-induced neuroinflammatory priming. First, we will provide a background on the basic principles of neuroimmunology. Next, we will discuss evidence that neuroinflammatory responses become primed in the context of both stress and aging. We will also describe cell-specific contributions to neuroinflammatory priming with a focus on microglia. Finally, common mechanisms underlying priming in the context of stress and aging will be discussed: these mechanisms include glucocorticoid signaling; accumulation of danger signals; dis-inhibition of microglia; and breakdown of circadian rhythms. Overall, there are multifarious parallels between stress- and aging-elicited neuroinflammatory priming, suggesting that stress may promote a form of premature aging. Further unravelling mechanisms underlying priming could lead to improved treatments for buffering against stress- and aging-elicited behavioral pathologies.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
References
-
- Agalave NM, Larsson M, Abdelmoaty S, Su J, Baharpoor A, Lundback P, Palmblad K, Andersson U, Harris H, Svensson CI, 2014. Spinal HMGB1 induces TLR4-mediated long-lasting hypersensitivity and glial activation and regulates pain-like behavior in experimental arthritis. Pain 155, 1802–1813. - PubMed
-
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM, 2007. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nature neuroscience 10, 1538–1543. - PubMed
-
- Alliot F, Godin I, Pessac B, 1999. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res 117, 145–152. - PubMed
-
- Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, Richardson P, Tipton T, Chapman MA, Riecken K, Beccari S, Sierra A, Molnar Z, Cragg MS, Garaschuk O, Perry VH, Gomez-Nicola D, 2017. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep 18, 391–405. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
