

Gene expression drives the evolution of dominance
- PMID: 30013096
- PMCID: PMC6048131
- DOI: 10.1038/s41467-018-05281-7
Gene expression drives the evolution of dominance
Abstract
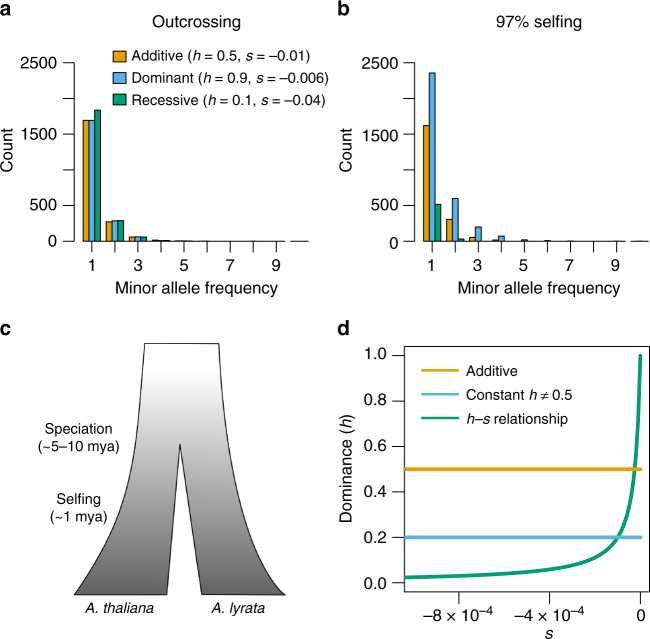
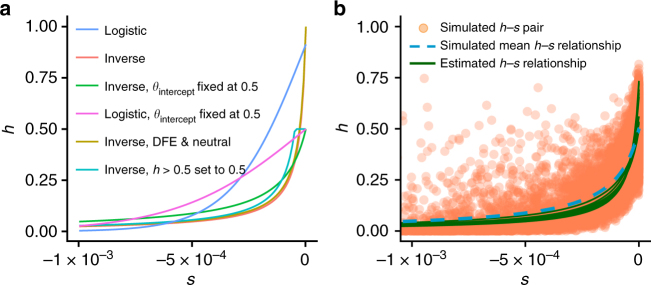
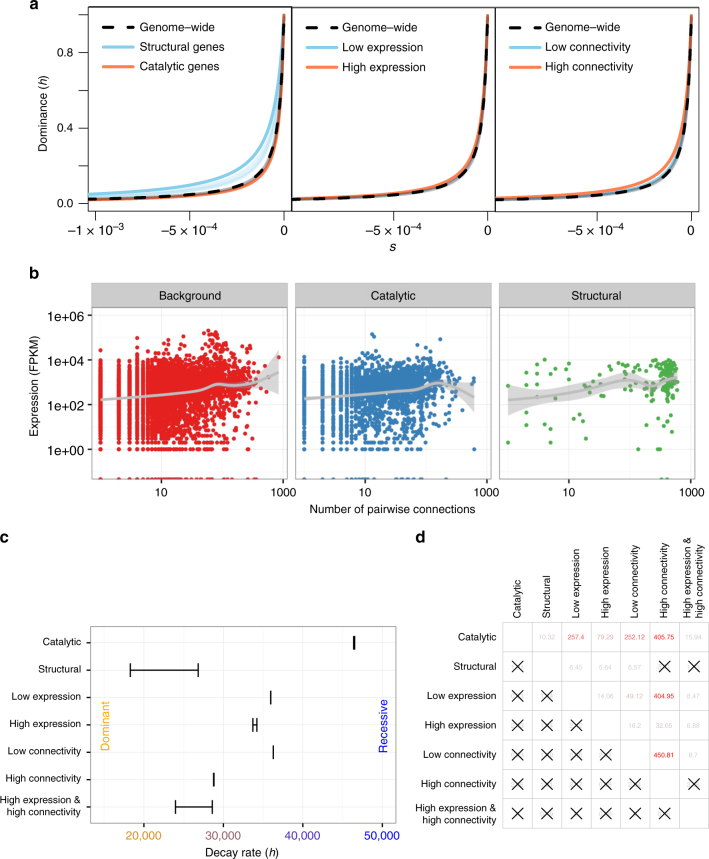
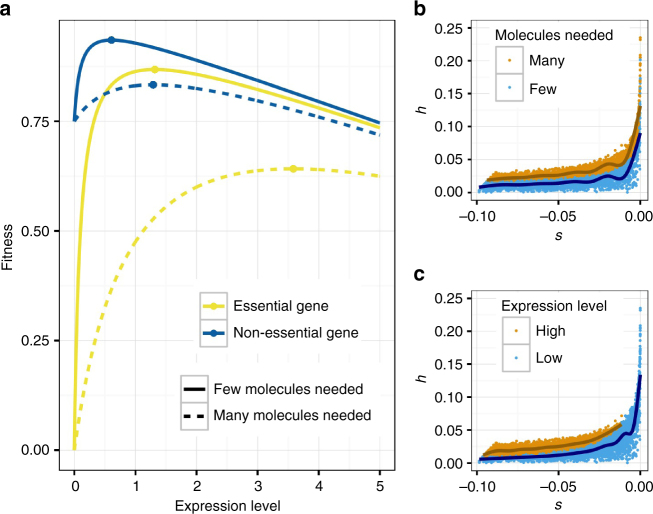
Dominance is a fundamental concept in molecular genetics and has implications for understanding patterns of genetic variation, evolution, and complex traits. However, despite its importance, the degree of dominance in natural populations is poorly quantified. Here, we leverage multiple mating systems in natural populations of Arabidopsis to co-estimate the distribution of fitness effects and dominance coefficients of new amino acid changing mutations. We find that more deleterious mutations are more likely to be recessive than less deleterious mutations. Further, this pattern holds across gene categories, but varies with the connectivity and expression patterns of genes. Our work argues that dominance arises as a consequence of the functional importance of genes and their optimal expression levels.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Fisher RA. The possible modification of the response of the wild type to recurrent mutations. Am. Nat. 1928;62:115–126. doi: 10.1086/280193. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
