

Genetics of trans-regulatory variation in gene expression
- PMID: 30014850
- PMCID: PMC6072440
- DOI: 10.7554/eLife.35471
Genetics of trans-regulatory variation in gene expression
Abstract
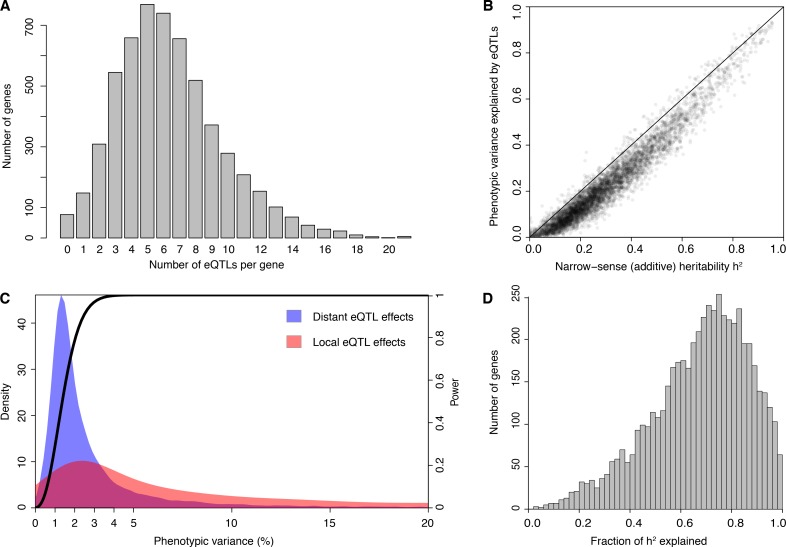
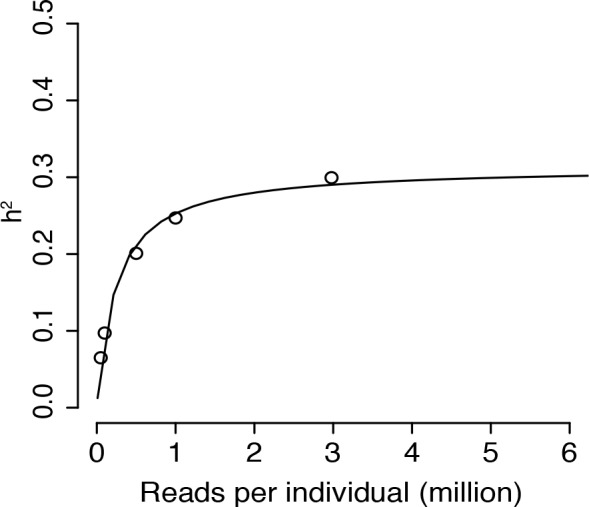
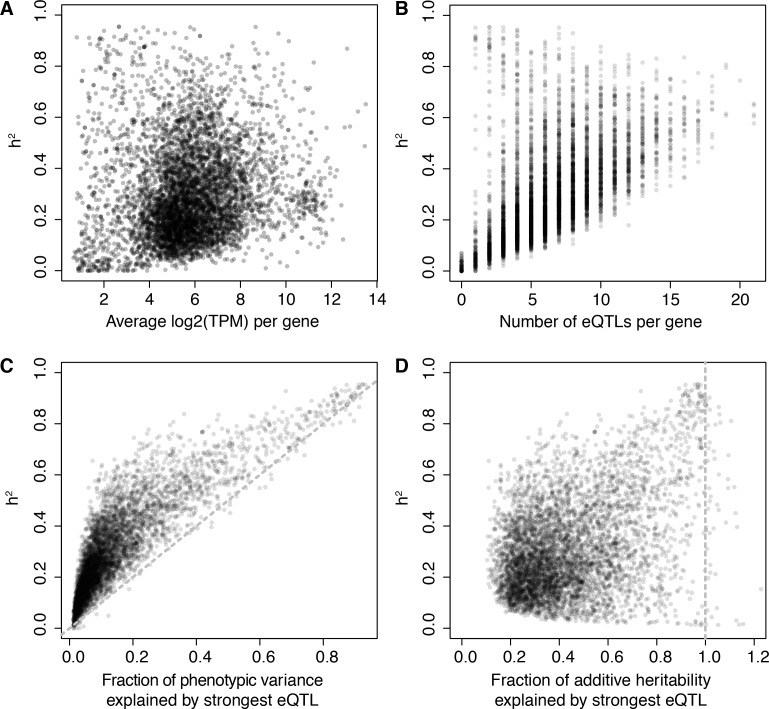
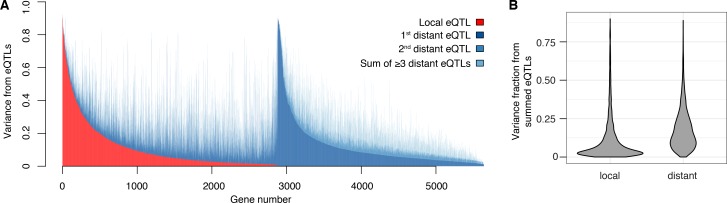
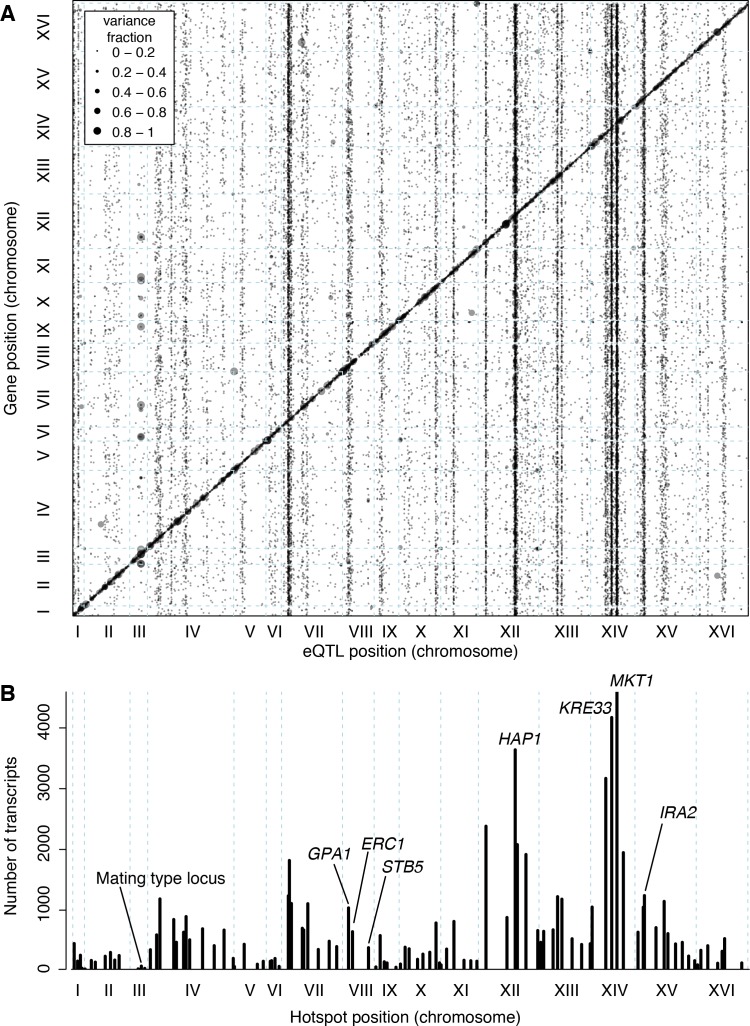
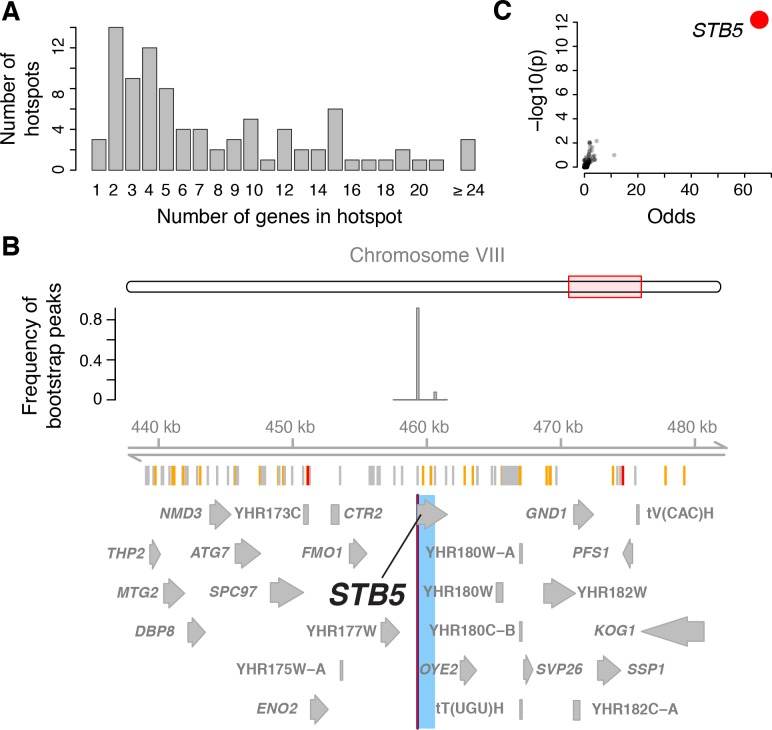
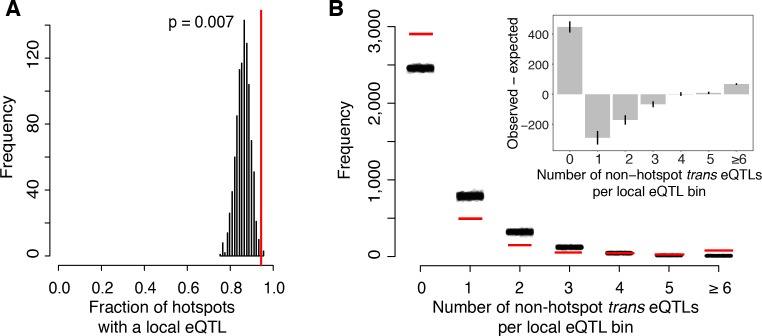
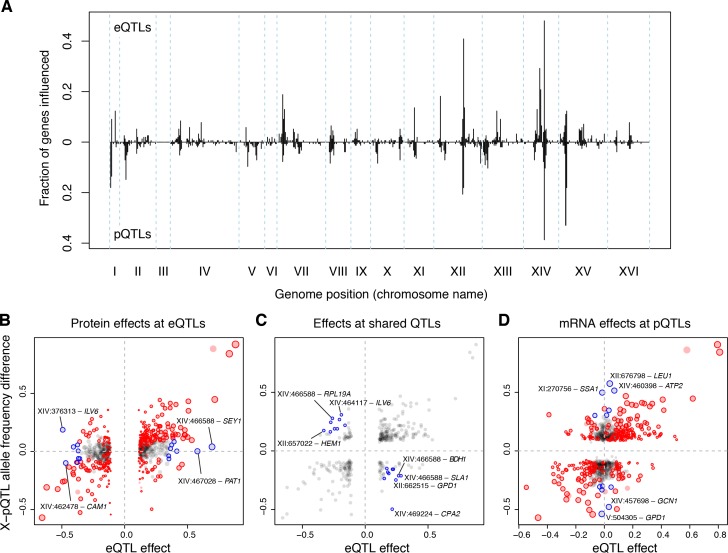
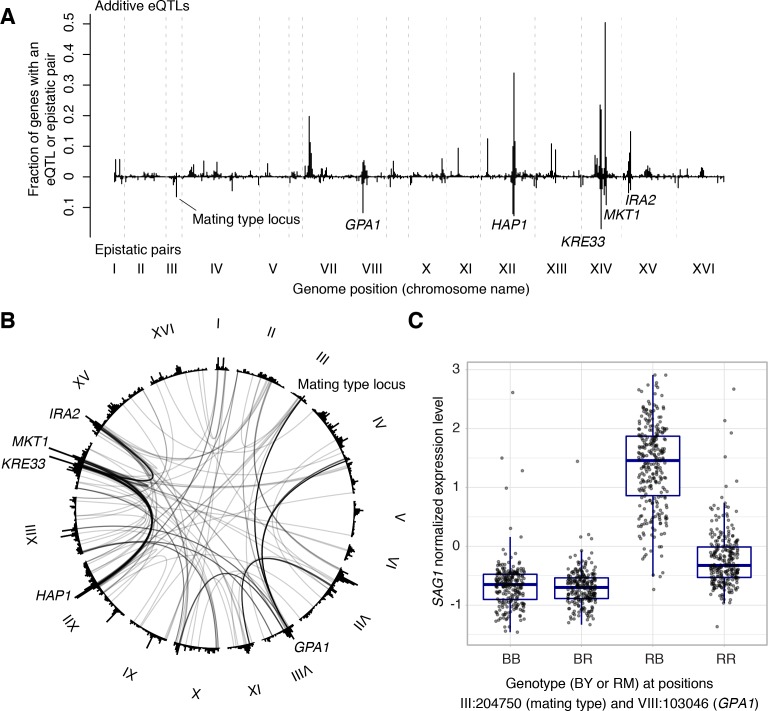
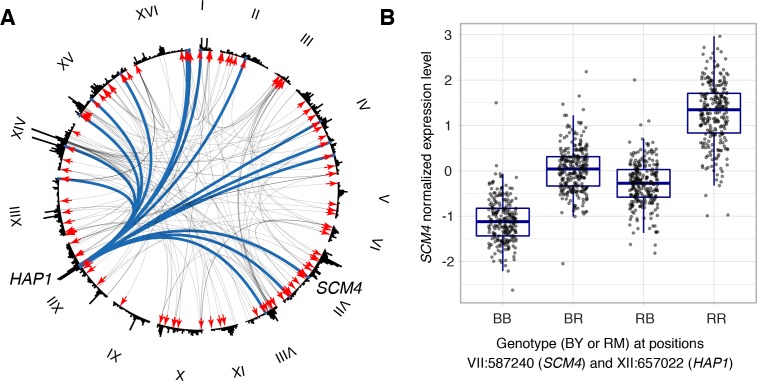
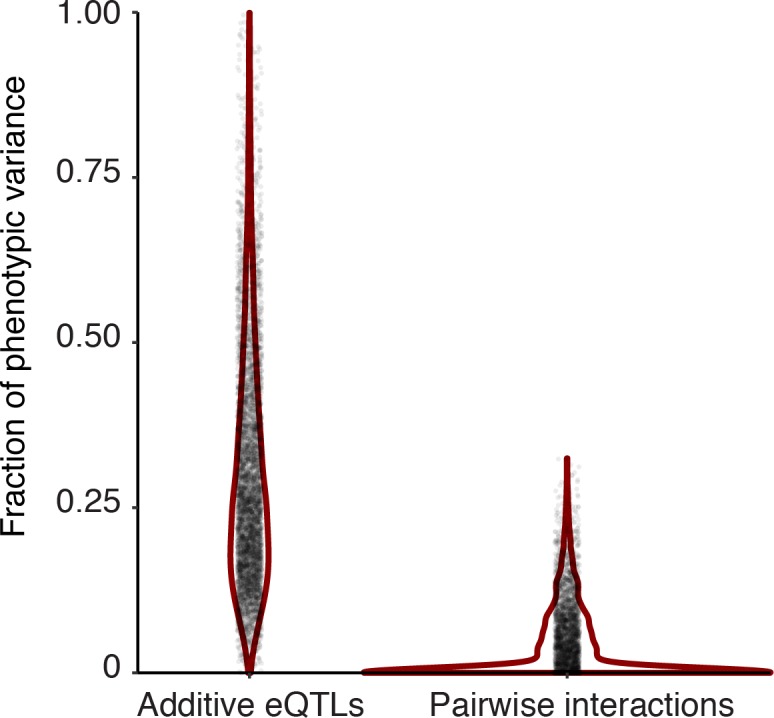
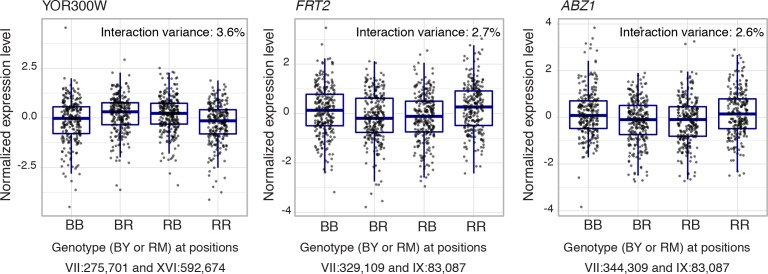
Heritable variation in gene expression forms a crucial bridge between genomic variation and the biology of many traits. However, most expression quantitative trait loci (eQTLs) remain unidentified. We mapped eQTLs by transcriptome sequencing in 1012 yeast segregants. The resulting eQTLs accounted for over 70% of the heritability of mRNA levels, allowing comprehensive dissection of regulatory variation. Most genes had multiple eQTLs. Most expression variation arose from trans-acting eQTLs distant from their target genes. Nearly all trans-eQTLs clustered at 102 hotspot locations, some of which influenced the expression of thousands of genes. Fine-mapped hotspot regions were enriched for transcription factor genes. While most genes had a local eQTL, most of these had no detectable effects on the expression of other genes in trans. Hundreds of non-additive genetic interactions accounted for small fractions of expression variation. These results reveal the complexity of genetic influences on transcriptome variation in unprecedented depth and detail.
Keywords: S. cerevisiae; chromosomes; eQTL; gene expression; genetic variation; genetics; genomics; regulatory variation.
© 2018, Albert et al.
Conflict of interest statement
FA, JB, JS, LD No competing interests declared, LK Reviewing editor, eLife
Figures
References
-
- Aguet F, Brown AA, Castel SE, Davis JR, He Y, Jo B, Mohammadi P, Park Y, Parsana P, Segrè AV, Strober BJ, Zappala Z, Cummings BB, Gelfand ET, Hadley K, Huang KH, Lek M, Li X, Nedzel JL, Nguyen DY, Noble MS, Sullivan TJ, Tukiainen T, MacArthur DG, Getz G, Addington A, Guan P, Koester S, Little AR, Lockhart NC, Moore HM, Rao A, Struewing JP, Volpi S, Brigham LE, Hasz R, Hunter M, Johns C, Johnson M, Kopen G, Leinweber WF, Lonsdale JT, McDonald A, Mestichelli B, Myer K, Roe B, Salvatore M, Shad S, Thomas JA, Walters G, Washington M, Wheeler J, Bridge J, Foster BA, Gillard BM, Karasik E, Kumar R, Miklos M, Moser MT, Jewell SD, Montroy RG, Rohrer DC, Valley D, Mash DC, Davis DA, Sobin L, Barcus ME, Branton PA, Abell NS, Balliu B, Delaneau O, Frésard L, Gamazon ER, Garrido-Martín D, Gewirtz ADH, Gliner G, Gloudemans MJ, Han B, He AZ, Hormozdiari F, Li X, Liu B, Kang EY, McDowell IC, Ongen H, Palowitch JJ, Peterson CB, Quon G, Ripke S, Saha A, Shabalin AA, Shimko TC, Sul JH, Teran NA, Tsang EK, Zhang H, Zhou Y-H, Bustamante CD, Cox NJ, Guigó R, Kellis M, McCarthy MI, Conrad DF, Eskin E, Li G, Nobel AB, Sabatti C, Stranger BE, Wen X, Wright FA, Ardlie KG, Dermitzakis ET, Lappalainen T, Aguet F, Ardlie KG, Cummings BB, Gelfand ET, Getz G, Hadley K, Handsaker RE, Huang KH, Kashin S, Karczewski KJ, Lek M, Li X, MacArthur DG, Nedzel JL, Nguyen DT, Noble MS, Segrè AV, Trowbridge CA, Tukiainen T, Abell NS, Balliu B, Barshir R, Basha O, Battle A, Bogu GK, Brown A, Brown CD, Castel SE, Chen LS, Chiang C, Conrad DF, Cox NJ, Damani FN, Davis JR, Delaneau O, Dermitzakis ET, Engelhardt BE, Eskin E, Ferreira PG, Frésard L, Gamazon ER, Garrido-Martín D, Gewirtz ADH, Gliner G, Gloudemans MJ, Guigo R, Hall IM, Han B, He Y, Hormozdiari F, Howald C, Kyung Im H, Jo B, Yong Kang E, Kim Y, Kim-Hellmuth S, Lappalainen T, Li G, Li X, Liu B, Mangul S, McCarthy MI, McDowell IC, Mohammadi P, Monlong J, Montgomery SB, Muñoz-Aguirre M, Ndungu AW, Nicolae DL, Nobel AB, Oliva M, Ongen H, Palowitch JJ, Panousis N, Papasaikas P, Park Y, Parsana P, Payne AJ, Peterson CB, Quan J, Reverter F, Sabatti C, Saha A, Sammeth M, Shabalin AA, Sodaei R, Stephens M, Stranger BE, Strober BJ, Sul JH, Tsang EK, Urbut S, van de Bunt M, Wang G, Wen X, Wright FA, Xi HS, Yeger-Lotem E, Zappala Z, Zaugg JB, Zhou Y-H, Akey JM, Bates D, Chan J, Chen LS, Claussnitzer M, Demanelis K, Diegel M, Doherty JA, Feinberg AP, Fernando MS, Halow J, Hansen KD, Haugen E, Hickey PF, Hou L, Jasmine F, Jian R, Jiang L, Johnson A, Kaul R, Kellis M, Kibriya MG, Lee K, Billy Li J, Li Q, Li X, Lin J, Lin S, Linder S, Linke C, Liu Y, Maurano MT, Molinie B, Montgomery SB, Nelson J, Neri FJ, Oliva M, Park Y, Pierce BL, Rinaldi NJ, Rizzardi LF, Sandstrom R, Skol A, Smith KS, Snyder MP, Stamatoyannopoulos J, Stranger BE, Tang H, Tsang EK, Wang L, Wang M, Van Wittenberghe N, Wu F, Zhang R, Nierras CR, Branton PA, Carithers LJ, Guan P, Moore HM, Rao A, Vaught JB, Gould SE, Lockart NC, Martin C, Struewing JP, Volpi S, Addington AM, Koester SE, Little AR, Brigham LE, Hasz R, Hunter M, Johns C, Johnson M, Kopen G, Leinweber WF, Lonsdale JT, McDonald A, Mestichelli B, Myer K, Roe B, Salvatore M, Shad S, Thomas JA, Walters G, Washington M, Wheeler J, Bridge J, Foster BA, Gillard BM, Karasik E, Kumar R, Miklos M, Moser MT, Jewell SD, Montroy RG, Rohrer DC, Valley DR, Davis DA, Mash DC, Undale AH, Smith AM, Tabor DE, Roche NV, McLean JA, Vatanian N, Robinson KL, Sobin L, Barcus ME, Valentino KM, Qi L, Hunter S, Hariharan P, Singh S, Um KS, Matose T, Tomaszewski MM, Barker LK, Mosavel M, Siminoff LA, Traino HM, Flicek P, Juettemann T, Ruffier M, Sheppard D, Taylor K, Trevanion SJ, Zerbino DR, Craft B, Goldman M, Haeussler M, Kent WJ, Lee CM, Paten B, Rosenbloom KR, Vivian J, Zhu J. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
