

Leukemogenic nucleophosmin mutation disrupts the transcription factor hub that regulates granulomonocytic fates
- PMID: 30015632
- PMCID: PMC6159976
- DOI: 10.1172/JCI97117
Leukemogenic nucleophosmin mutation disrupts the transcription factor hub that regulates granulomonocytic fates
Abstract
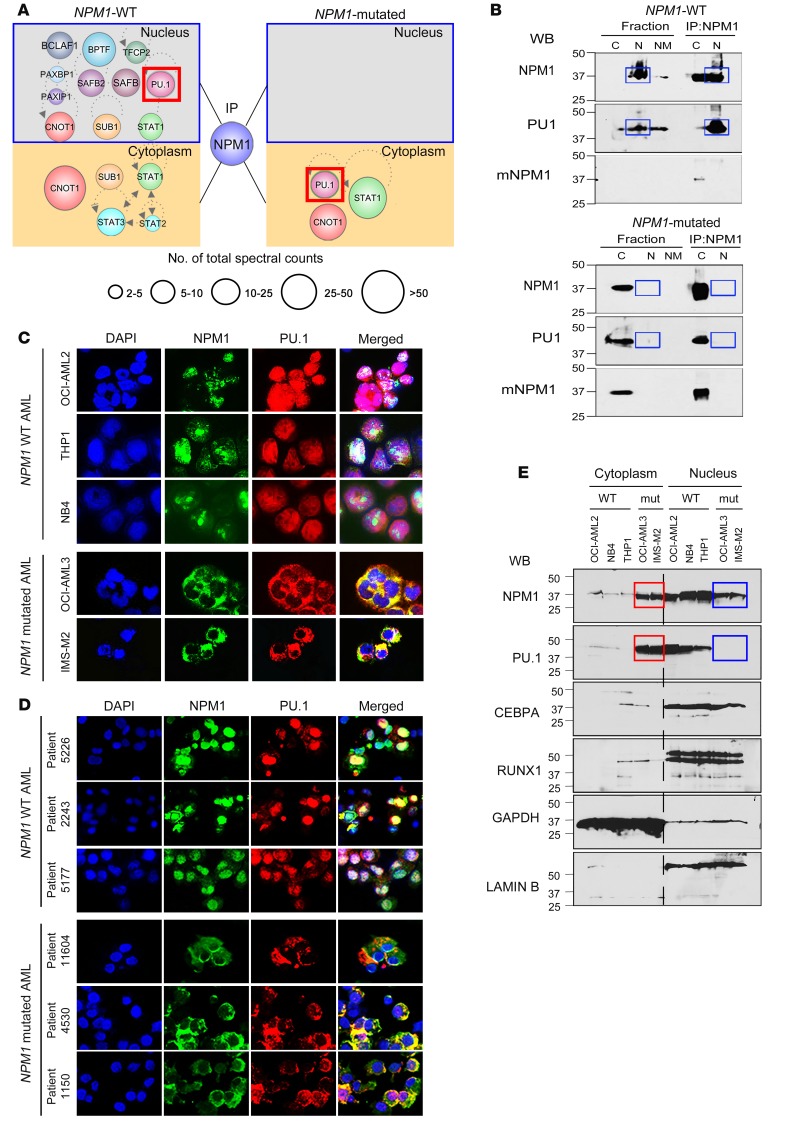
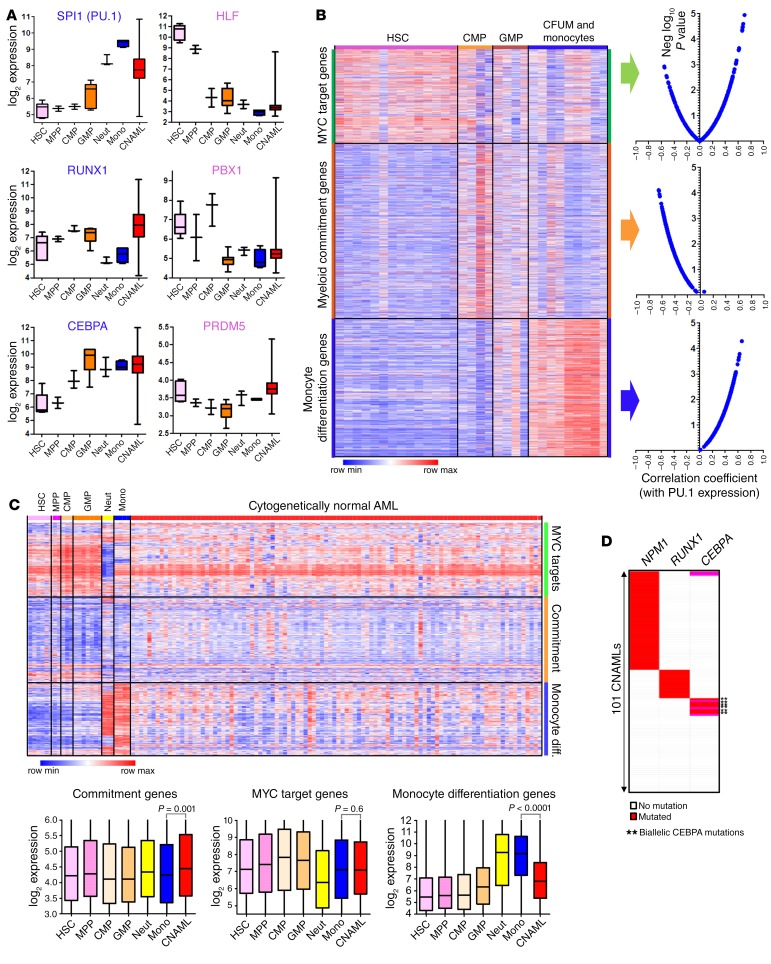
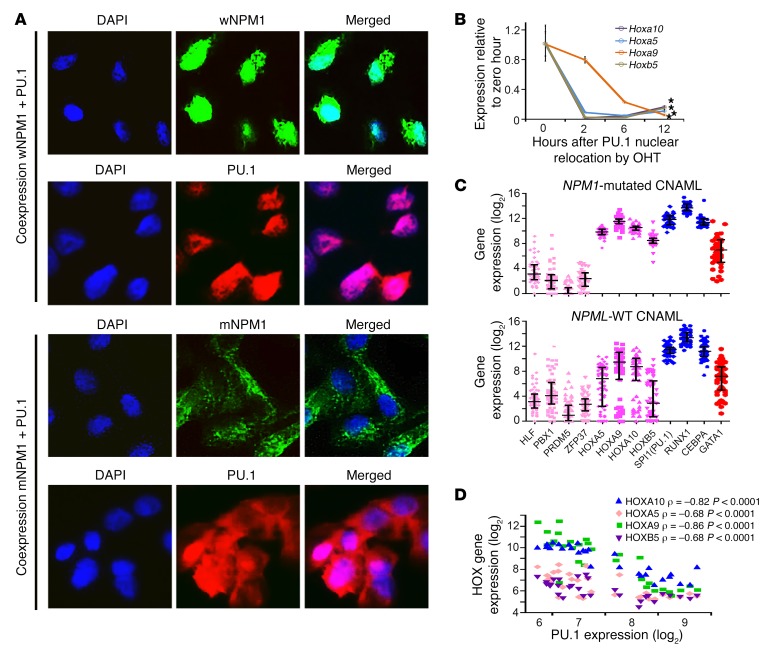
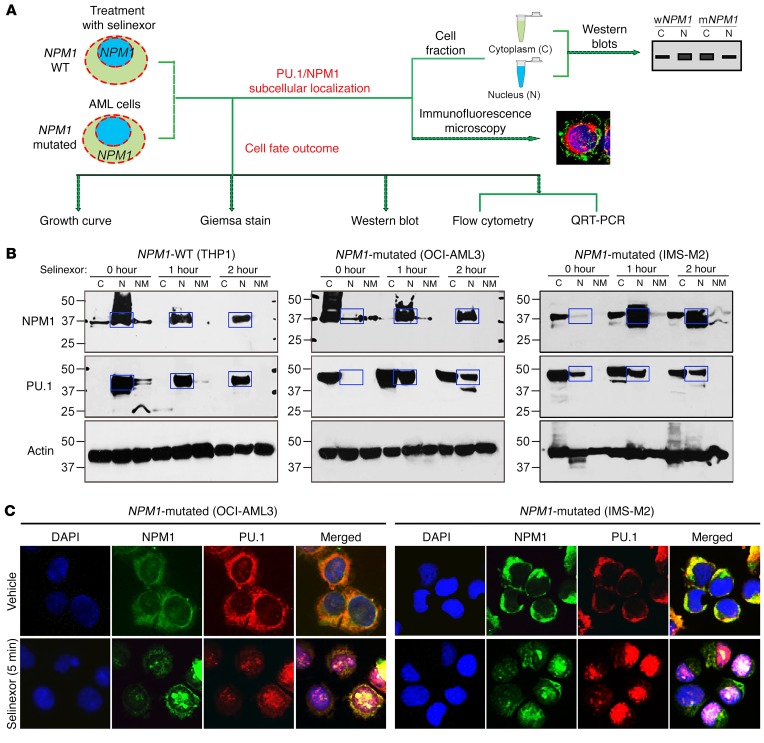
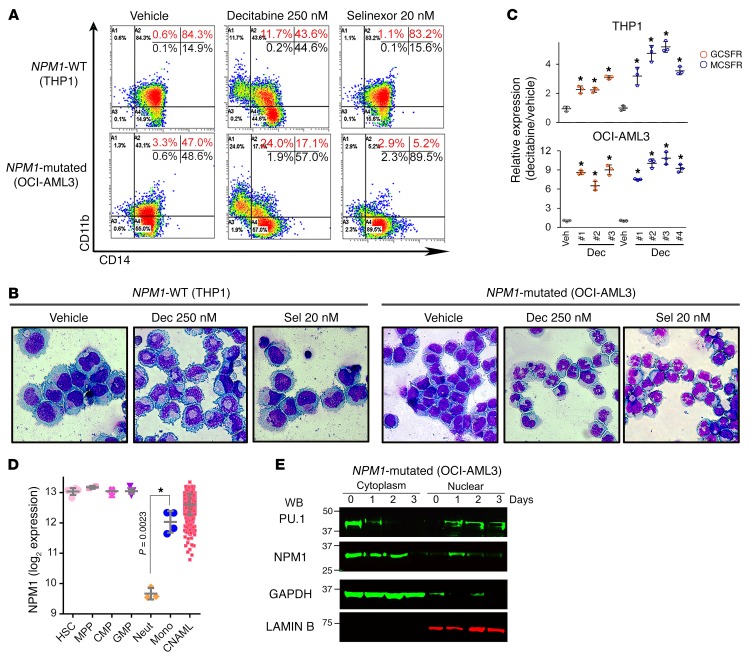
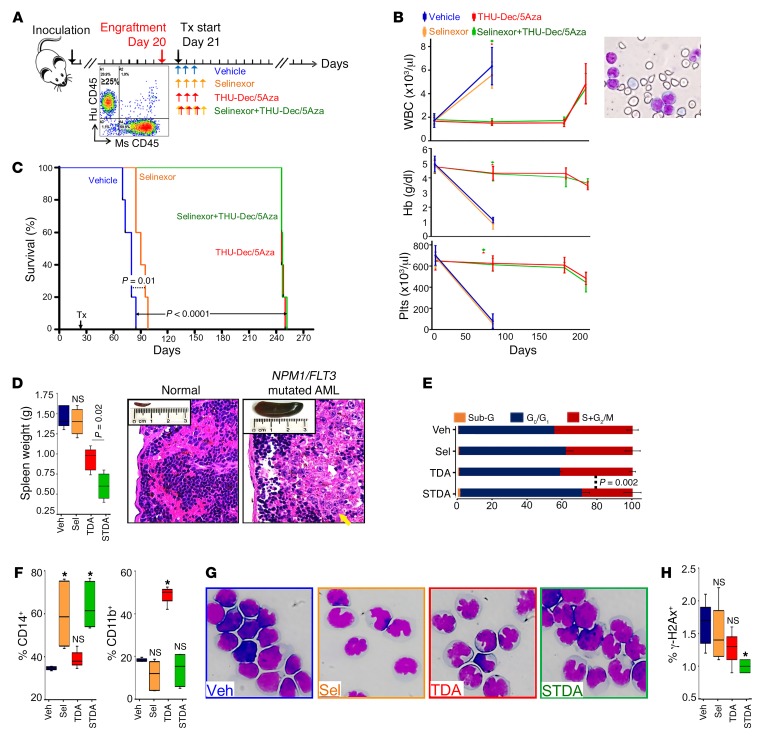
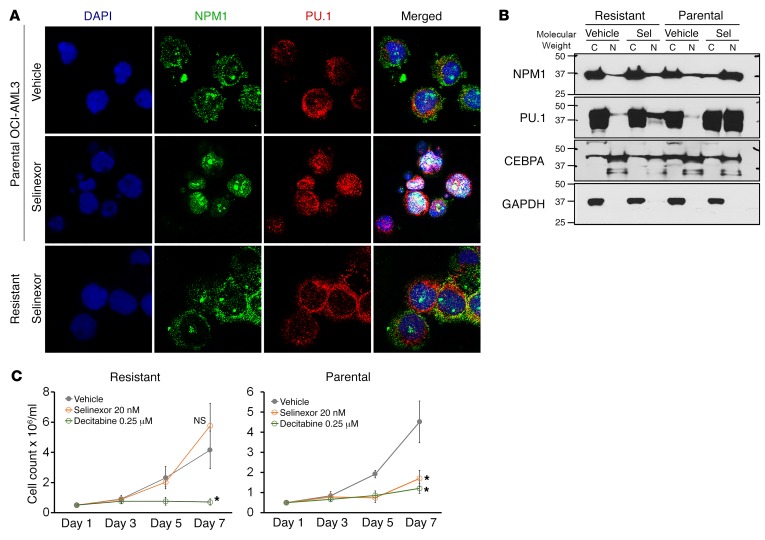
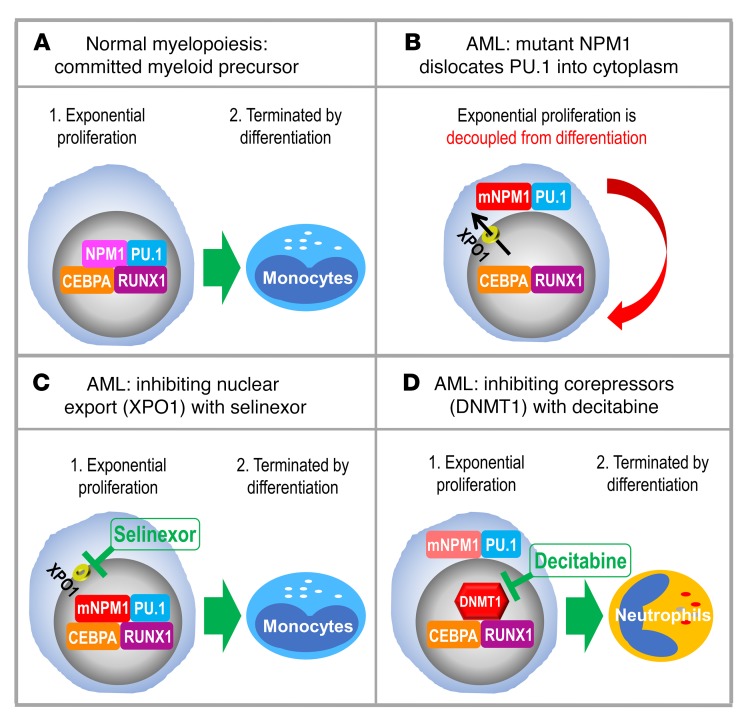
Nucleophosmin (NPM1) is among the most frequently mutated genes in acute myeloid leukemia (AML). It is not known, however, how the resulting oncoprotein mutant NPM1 is leukemogenic. To reveal the cellular machinery in which NPM1 participates in myeloid cells, we analyzed the endogenous NPM1 protein interactome by mass spectrometry and discovered abundant amounts of the master transcription factor driver of monocyte lineage differentiation PU.1 (also known as SPI1). Mutant NPM1, which aberrantly accumulates in cytoplasm, dislocated PU.1 into cytoplasm with it. CEBPA and RUNX1, the master transcription factors that collaborate with PU.1 to activate granulomonocytic lineage fates, remained nuclear; but without PU.1, their coregulator interactions were toggled from coactivators to corepressors, repressing instead of activating more than 500 granulocyte and monocyte terminal differentiation genes. An inhibitor of nuclear export, selinexor, by locking mutant NPM1/PU.1 in the nucleus, activated terminal monocytic fates. Direct depletion of the corepressor DNA methyltransferase 1 (DNMT1) from the CEBPA/RUNX1 protein interactome using the clinical drug decitabine activated terminal granulocytic fates. Together, these noncytotoxic treatments extended survival by more than 160 days versus vehicle in a patient-derived xenotransplant model of NPM1/FLT3-mutated AML. In sum, mutant NPM1 represses monocyte and granulocyte terminal differentiation by disrupting PU.1/CEBPA/RUNX1 collaboration, a transforming action that can be reversed by pharmacodynamically directed dosing of clinical small molecules.
Keywords: Epigenetics; Hematology; Leukemias; Oncology; Transport.
Conflict of interest statement
Figures
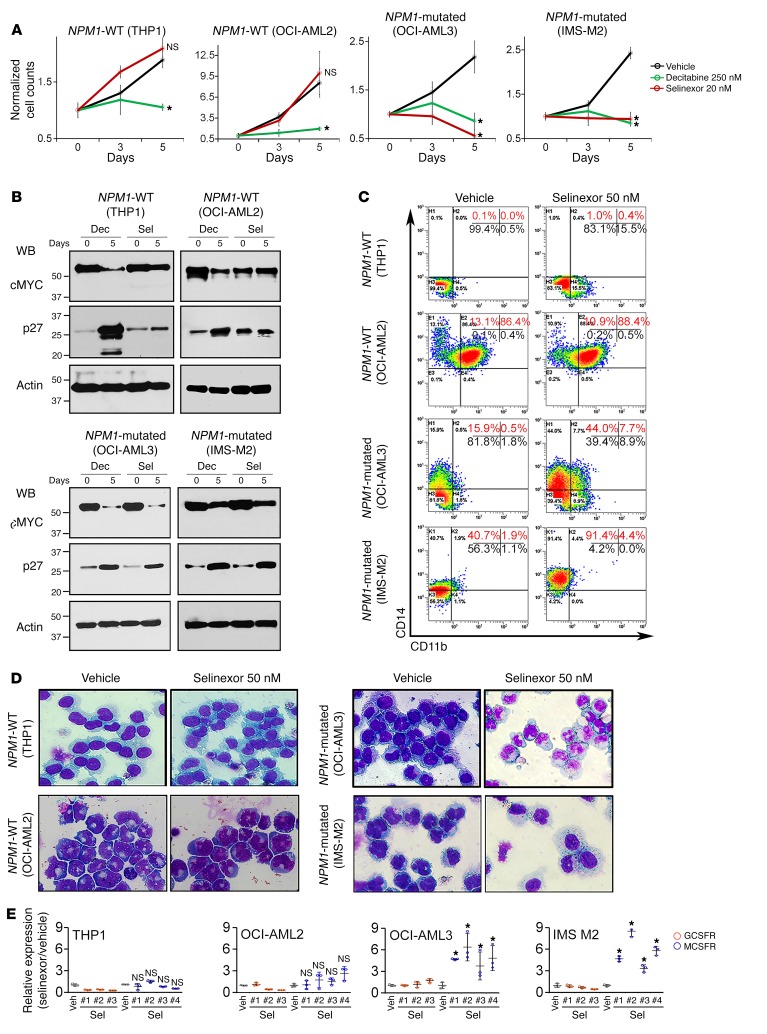
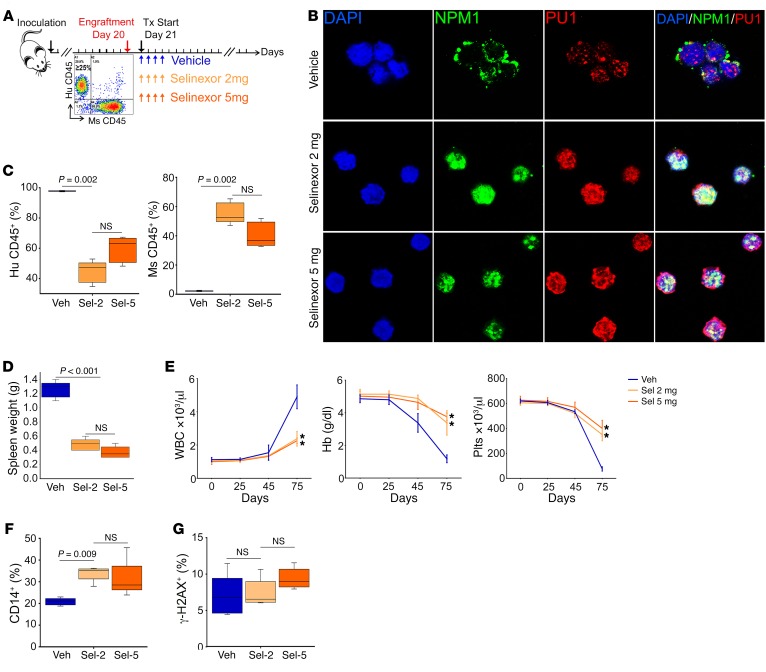
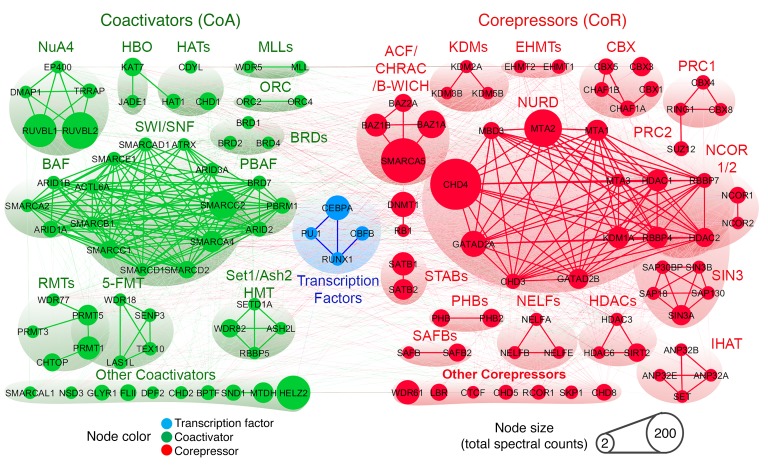
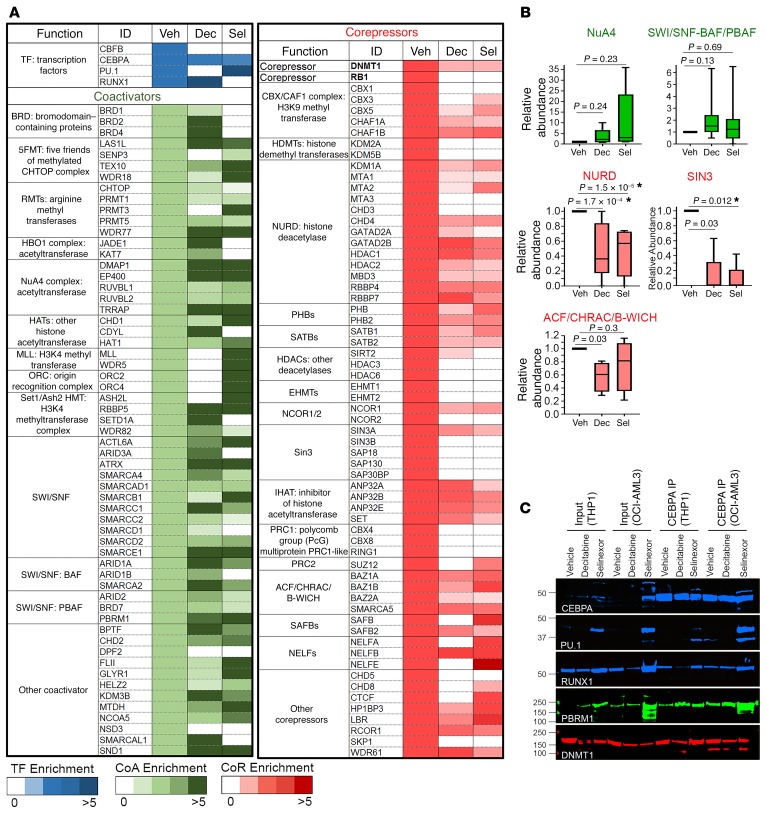
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
