

Mavacamten stabilizes an autoinhibited state of two-headed cardiac myosin
- PMID: 30018063
- PMCID: PMC6094135
- DOI: 10.1073/pnas.1720342115
Mavacamten stabilizes an autoinhibited state of two-headed cardiac myosin
Abstract
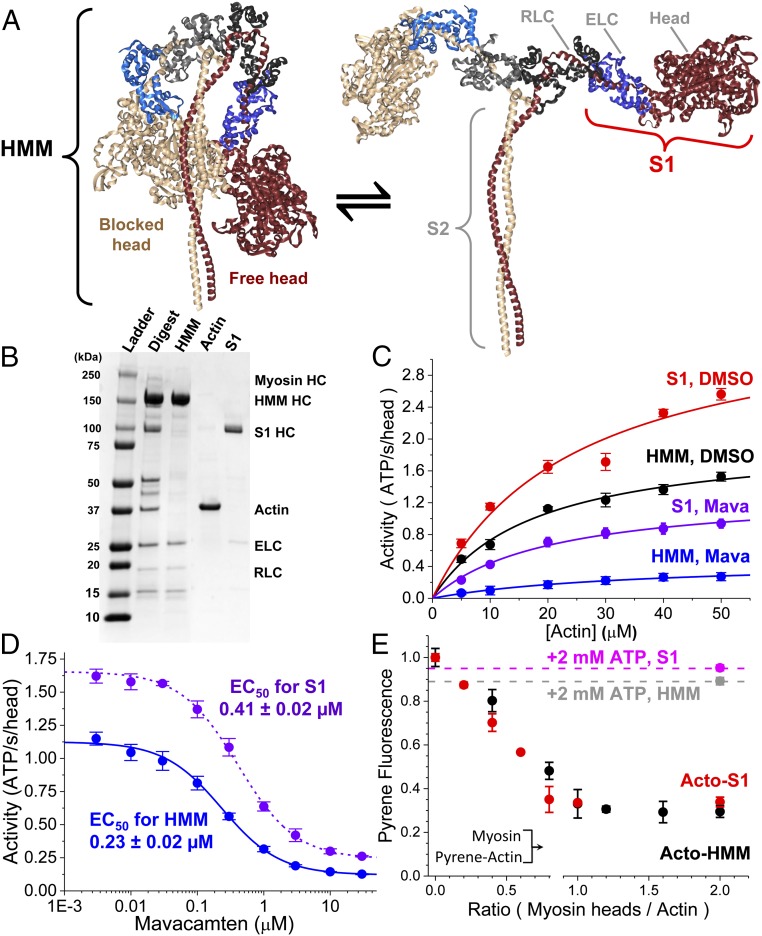
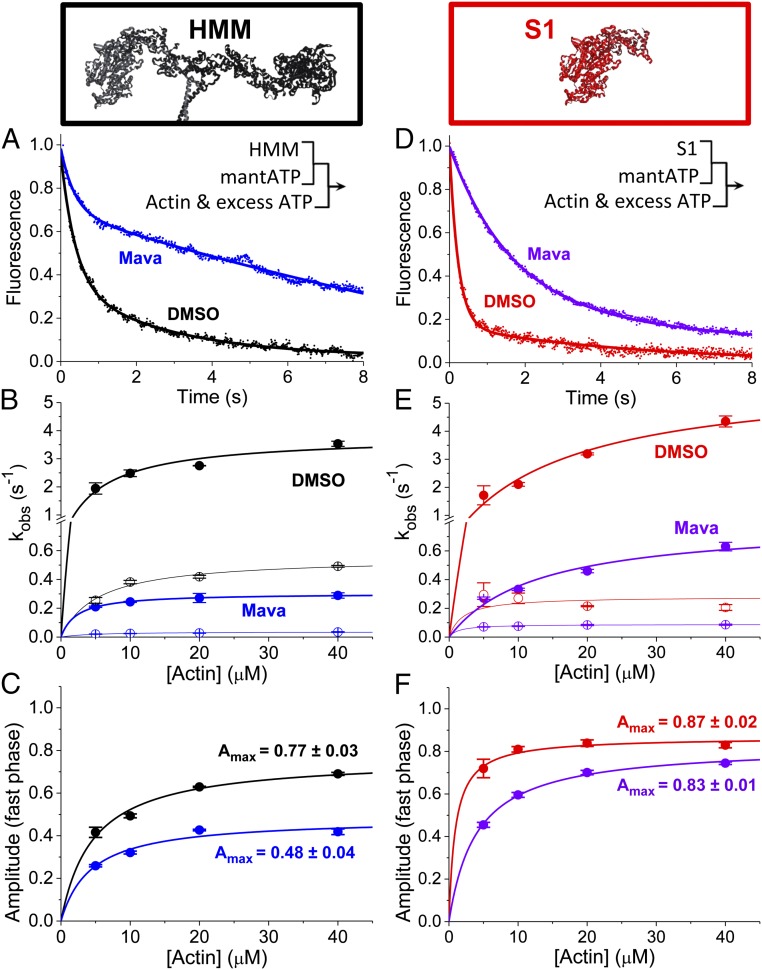
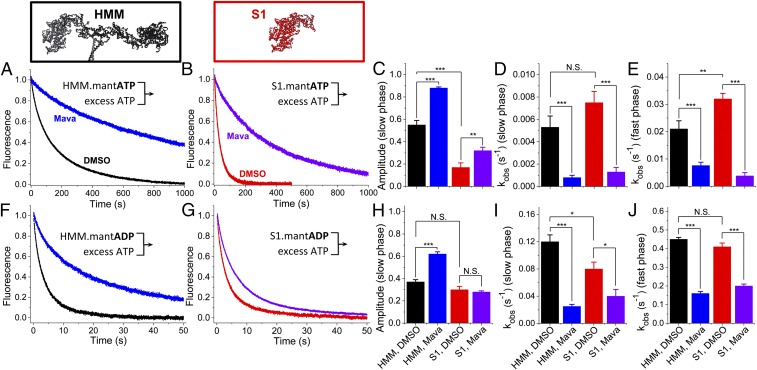
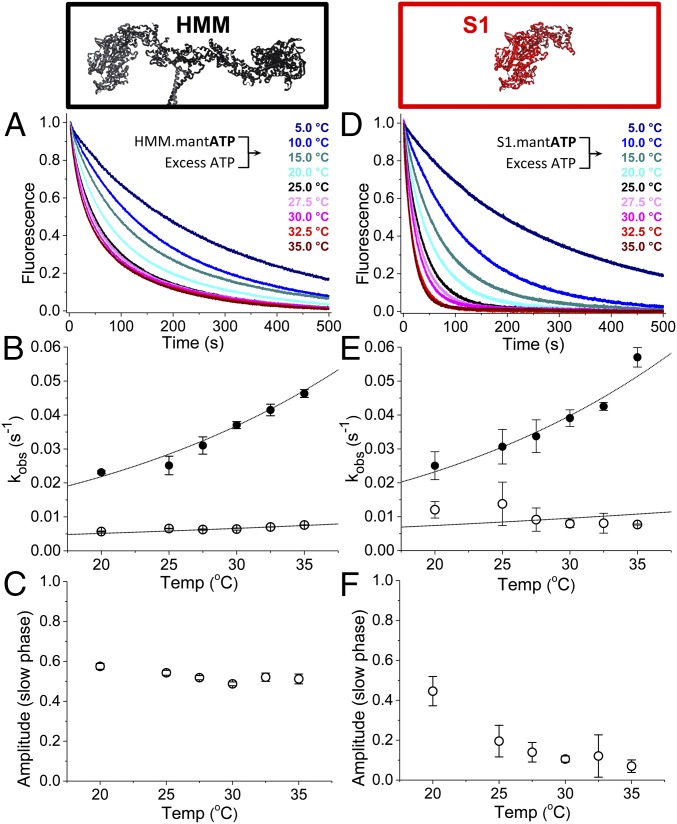
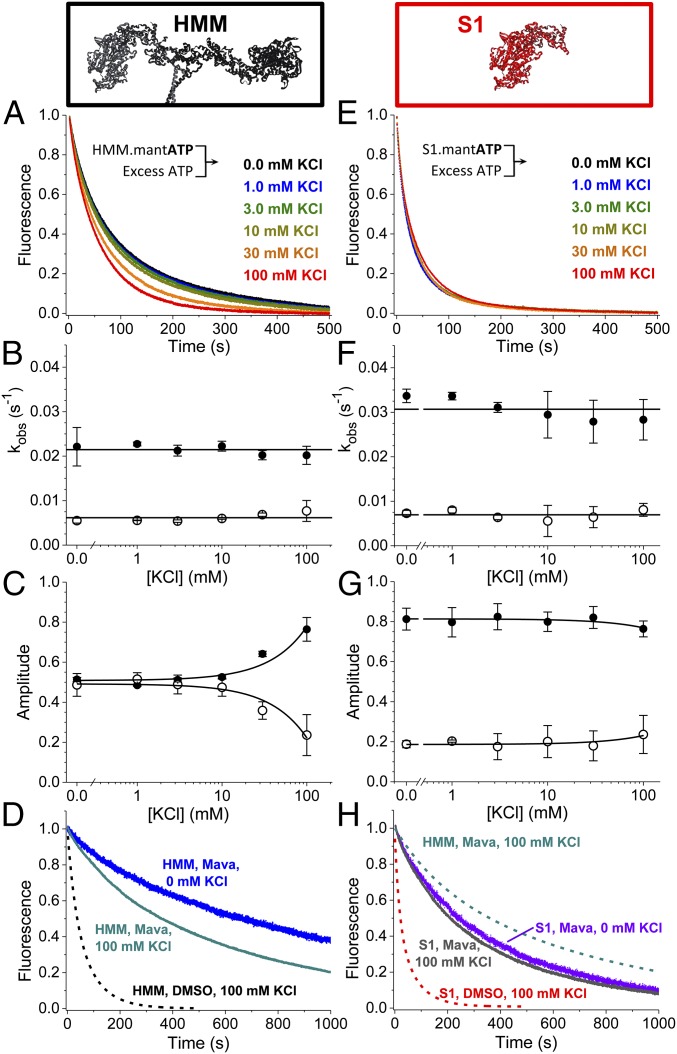
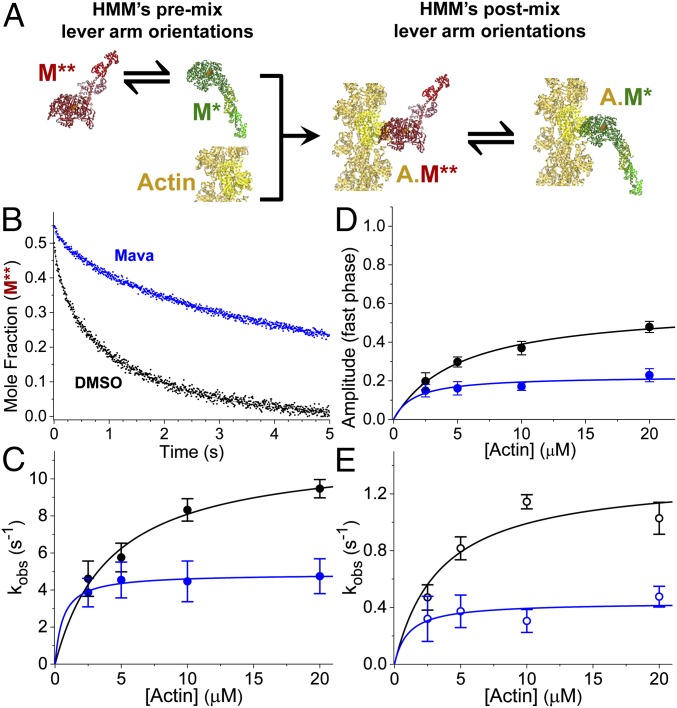
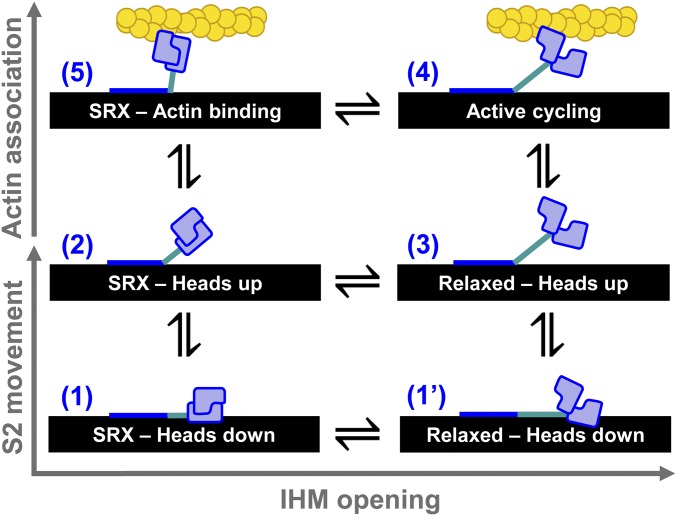
We used transient biochemical and structural kinetics to elucidate the molecular mechanism of mavacamten, an allosteric cardiac myosin inhibitor and a prospective treatment for hypertrophic cardiomyopathy. We find that mavacamten stabilizes an autoinhibited state of two-headed cardiac myosin not found in the single-headed S1 myosin motor fragment. We determined this by measuring cardiac myosin actin-activated and actin-independent ATPase and single-ATP turnover kinetics. A two-headed myosin fragment exhibits distinct autoinhibited ATP turnover kinetics compared with a single-headed fragment. Mavacamten enhanced this autoinhibition. It also enhanced autoinhibition of ADP release. Furthermore, actin changes the structure of the autoinhibited state by forcing myosin lever-arm rotation. Mavacamten slows this rotation in two-headed myosin but does not prevent it. We conclude that cardiac myosin is regulated in solution by an interaction between its two heads and propose that mavacamten stabilizes this state.
Keywords: allosteric inhibitor; cardiac myosin; hypertrophic cardiomyopathy; mavacamten; superrelaxed state.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Maron BJ, et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–789. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
