

Plasminogen activation triggers transthyretin amyloidogenesis in vitro
- PMID: 30018138
- PMCID: PMC6139548
- DOI: 10.1074/jbc.RA118.003990
Plasminogen activation triggers transthyretin amyloidogenesis in vitro
Abstract
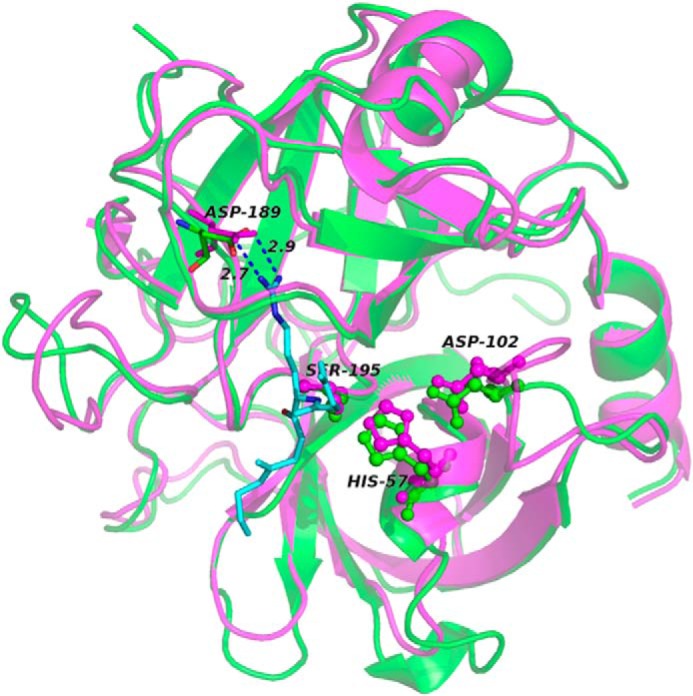
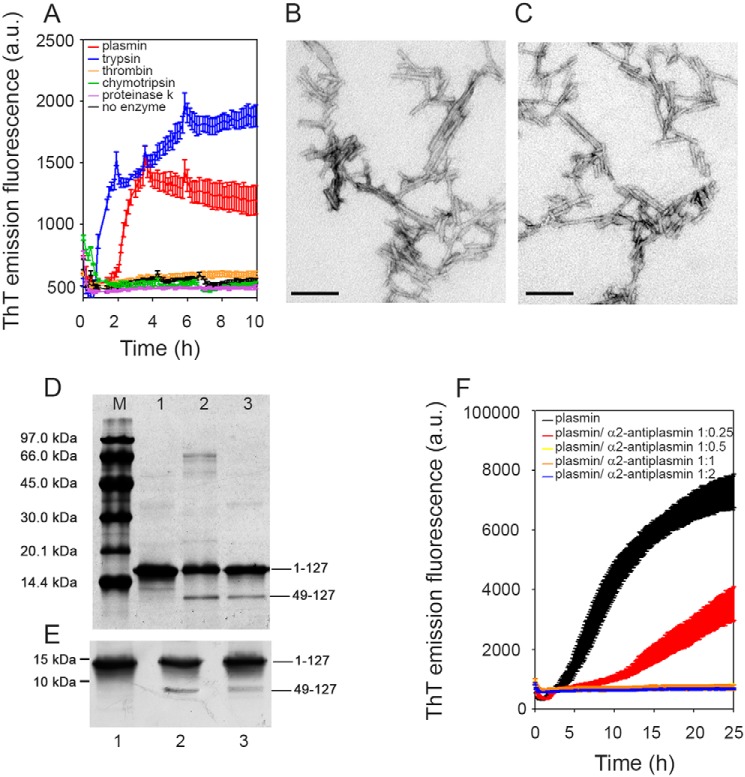
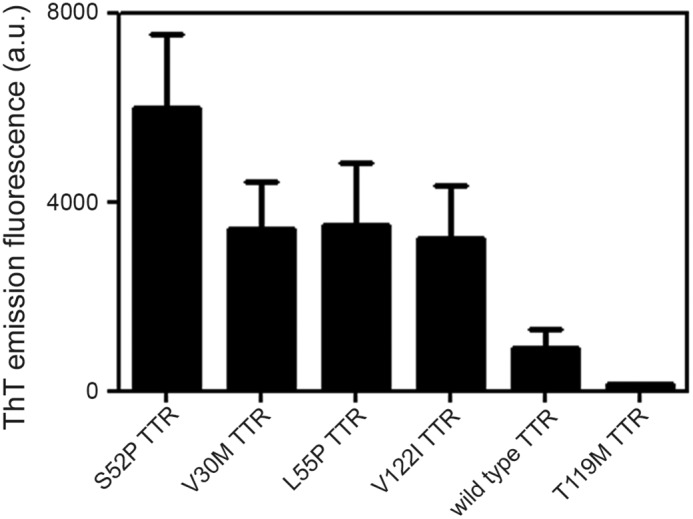
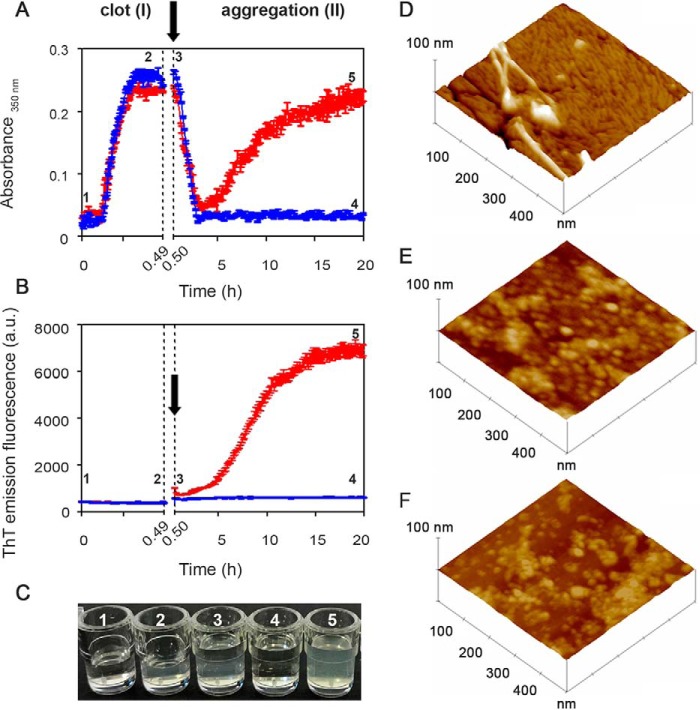
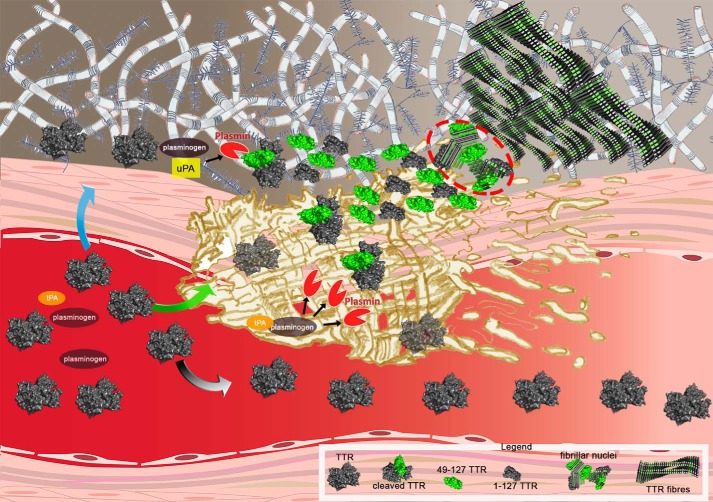
Systemic amyloidosis is a usually fatal disease caused by extracellular accumulation of abnormal protein fibers, amyloid fibrils, derived by misfolding and aggregation of soluble globular plasma protein precursors. Both WT and genetic variants of the normal plasma protein transthyretin (TTR) form amyloid, but neither the misfolding leading to fibrillogenesis nor the anatomical localization of TTR amyloid deposition are understood. We have previously shown that, under physiological conditions, trypsin cleaves human TTR in a mechano-enzymatic mechanism that generates abundant amyloid fibrils in vitro In sharp contrast, the widely used in vitro model of denaturation and aggregation of TTR by prolonged exposure to pH 4.0 yields almost no clearly defined amyloid fibrils. However, the exclusive duodenal location of trypsin means that this enzyme cannot contribute to systemic extracellular TTR amyloid deposition in vivo Here, we therefore conducted a bioinformatics search for systemically active tryptic proteases with appropriate tissue distribution, which unexpectedly identified plasmin as the leading candidate. We confirmed that plasmin, just as trypsin, selectively cleaves human TTR between residues 48 and 49 under physiological conditions in vitro Truncated and full-length protomers are then released from the native homotetramer and rapidly aggregate into abundant fibrils indistinguishable from ex vivo TTR amyloid. Our findings suggest that physiological fibrinolysis is likely to play a critical role in TTR amyloid formation in vivo Identification of this surprising intersection between two hitherto unrelated pathways opens new avenues for elucidating the mechanisms of TTR amyloidosis, for seeking susceptibility risk factors, and for therapeutic innovation.
Keywords: amyloid; amyloid fibrillogenesis; amyloidogenesis; fibril; mechano-enzymatic mechanism; protease; protein aggregation; systemic amyloidosis; tissue plasminogen activator (tPA); transthyretin.
© 2018 Mangione et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Mangione P. P., Porcari R., Gillmore J. D., Pucci P., Monti M., Porcari M., Giorgetti S., Marchese L., Raimondi S., Serpell L. C., Chen W., Relini A., Marcoux J., Clatworthy I. R., Taylor G. W., et al. (2014) Proteolytic cleavage of Ser52Pro variant transthyretin triggers its amyloid fibrillogenesis. Proc. Natl. Acad. Sci. U.S.A. 111, 1539–1544 10.1073/pnas.1317488111 - DOI - PMC - PubMed
-
- Klimtchuk E. S., Prokaeva T., Frame N. M., Abdullahi H. A., Spencer B., Dasari S., Cui H., Berk J. L., Kurtin P. J., Connors L. H., and Gursky O. (2018) Unusual duplication mutation in a surface loop of human transthyretin leads to an aggressive drug-resistant amyloid disease. Proc. Natl. Acad. Sci. U.S.A. 115, E6428–E6436 10.1073/pnas.1802977115 - DOI - PMC - PubMed
-
- Marcoux J., Mangione P. P., Porcari R., Degiacomi M. T., Verona G., Taylor G. W., Giorgetti S., Raimondi S., Sanglier-Cianferani S., Benesch J. L., Cecconi C., Naqvi M. M., Gillmore J. D., Hawkins P. N., Stoppini M., Robinson C. V., Pepys M. B., and Bellotti V. (2015) A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol. Med. 7, 1337–1349 10.15252/emmm.201505357 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
