

SECAPR-a bioinformatics pipeline for the rapid and user-friendly processing of targeted enriched Illumina sequences, from raw reads to alignments
- PMID: 30023140
- PMCID: PMC6047508
- DOI: 10.7717/peerj.5175
SECAPR-a bioinformatics pipeline for the rapid and user-friendly processing of targeted enriched Illumina sequences, from raw reads to alignments
Abstract
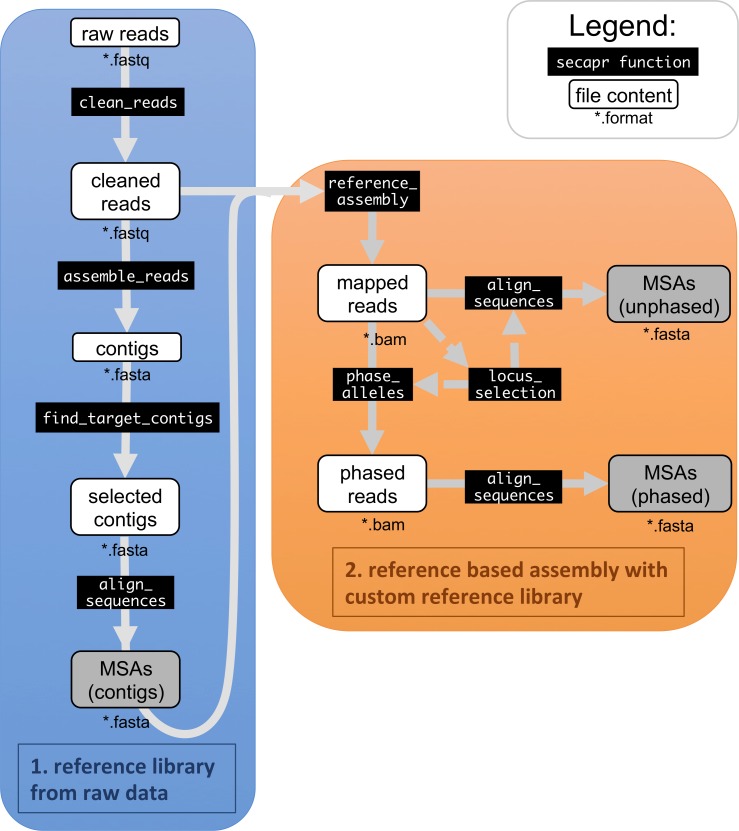
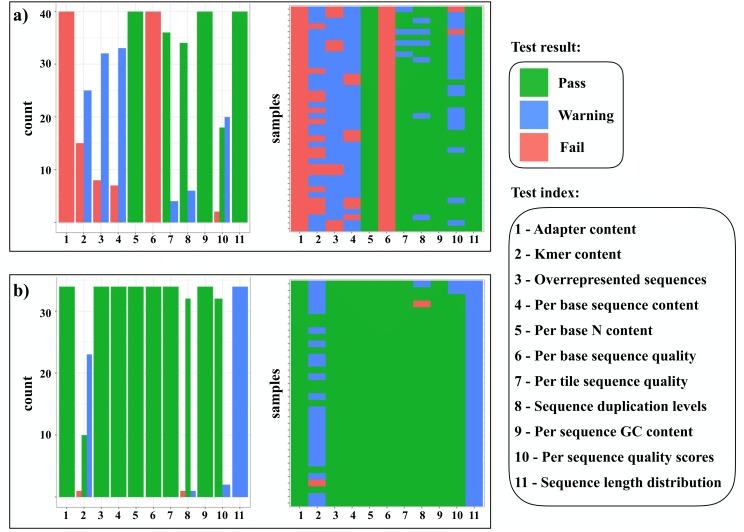
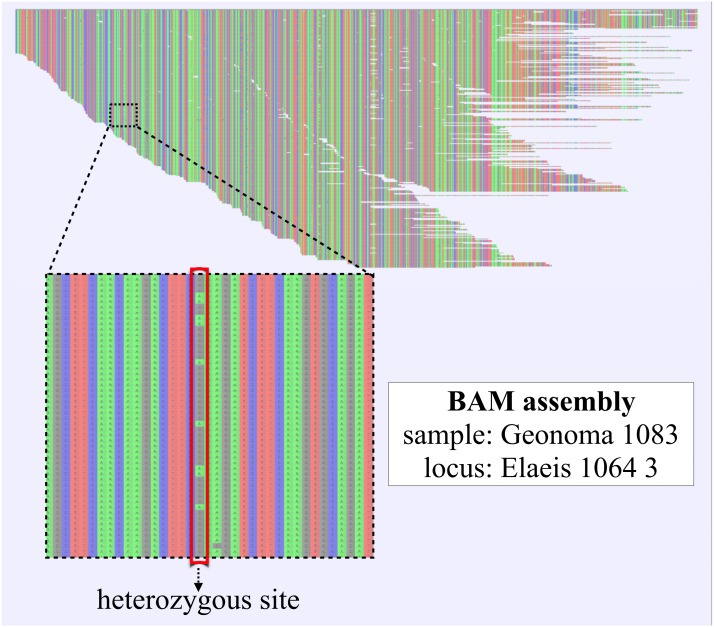
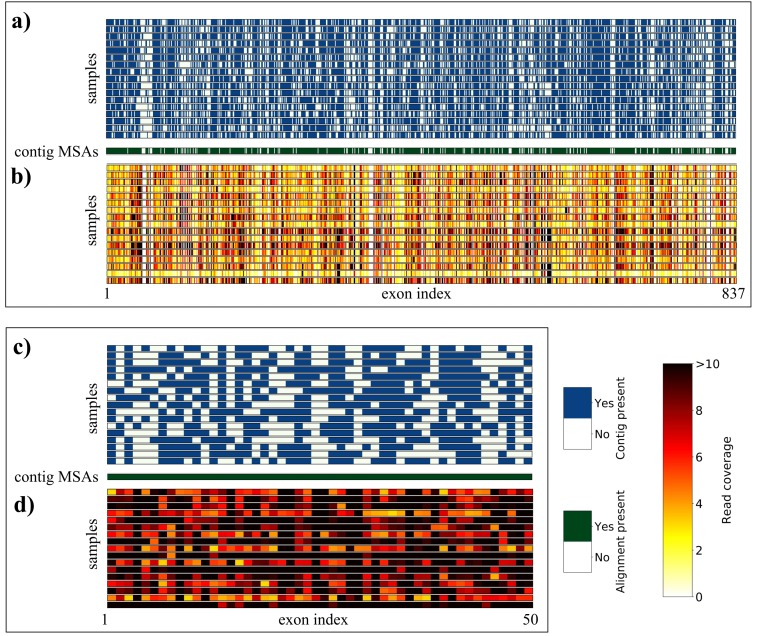
Evolutionary biology has entered an era of unprecedented amounts of DNA sequence data, as new sequencing technologies such as Massive Parallel Sequencing (MPS) can generate billions of nucleotides within less than a day. The current bottleneck is how to efficiently handle, process, and analyze such large amounts of data in an automated and reproducible way. To tackle these challenges we introduce the Sequence Capture Processor (SECAPR) pipeline for processing raw sequencing data into multiple sequence alignments for downstream phylogenetic and phylogeographic analyses. SECAPR is user-friendly and we provide an exhaustive empirical data tutorial intended for users with no prior experience with analyzing MPS output. SECAPR is particularly useful for the processing of sequence capture (synonyms: target or hybrid enrichment) datasets for non-model organisms, as we demonstrate using an empirical sequence capture dataset of the palm genus Geonoma (Arecaceae). Various quality control and plotting functions help the user to decide on the most suitable settings for even challenging datasets. SECAPR is an easy-to-use, free, and versatile pipeline, aimed to enable efficient and reproducible processing of MPS data for many samples in parallel.
Keywords: Allele phasing; Assembly; BAM; Contig; Exon capture; FASTQ; Next generation sequencing (NGS); Phylogenetics; Phylogeography; Target capture.
Conflict of interest statement
The authors declare there are no competing interests.
Figures
References
-
- Andermann T, Fernandes AM, Olsson U, Töpel M, Pfeil B, Oxelman B, Aleixo A, Faircloth BC, Antonelli A, Renner S. Allele phasing greatly improves the phylogenetic utility of ultraconserved elements. Systematic Biology. 2018 doi: 10.1093/sysbio/syy039. Epub ahead of print May 15 2018. - DOI - PMC - PubMed
-
- BabrahamBioinformatics FastQC a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [2 June]. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
- Botero-Castro F, Tilak MK, Justy F, Catzeflis F, Delsuc F, Douzery EJP. Next-generation sequencing and phylogenetic signal of complete mitochondrial genomes for resolving the evolutionary history of leaf-nosed bats (Phyllostomidae) Molecular Phylogenetics and Evolution. 2013;69:728–739. doi: 10.1016/j.ympev.2013.07.003. - DOI - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
