

Novel digenic inheritance of PCDH15 and USH1G underlies profound non-syndromic hearing impairment
- PMID: 30029624
- PMCID: PMC6053831
- DOI: 10.1186/s12881-018-0618-5
Novel digenic inheritance of PCDH15 and USH1G underlies profound non-syndromic hearing impairment
Abstract
Background: Digenic inheritance is the simplest model of oligenic disease. It can be observed when there is a strong epistatic interaction between two loci. For both syndromic and non-syndromic hearing impairment, several forms of digenic inheritance have been reported.
Methods: We performed exome sequencing in a Pakistani family with profound non-syndromic hereditary hearing impairment to identify the genetic cause of disease.
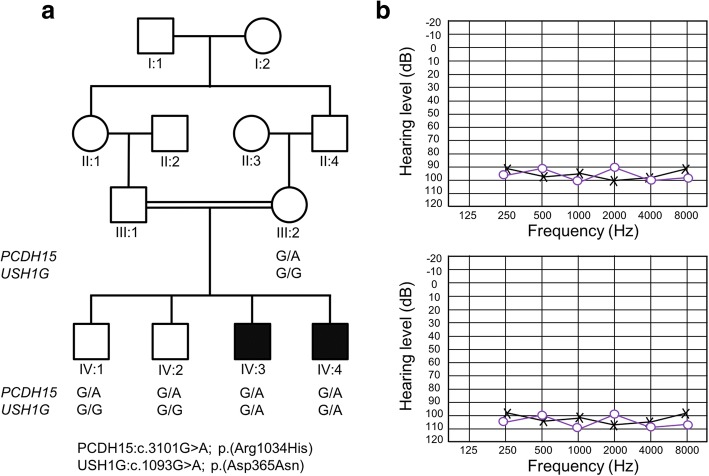
Results: We found that this family displays digenic inheritance for two trans heterozygous missense mutations, one in PCDH15 [p.(Arg1034His)] and another in USH1G [p.(Asp365Asn)]. Both of these genes are known to cause autosomal recessive non-syndromic hearing impairment and Usher syndrome. The protein products of PCDH15 and USH1G function together at the stereocilia tips in the hair cells and are necessary for proper mechanotransduction. Epistasis between Pcdh15 and Ush1G has been previously reported in digenic heterozygous mice. The digenic mice displayed a significant decrease in hearing compared to age-matched heterozygous animals. Until now no human examples have been reported.
Conclusions: The discovery of novel digenic inheritance mechanisms in hereditary hearing impairment will aid in understanding the interaction between defective proteins and further define inner ear function and its interactome.
Keywords: Deafness; Digenic inheritance; Hearing impairment; PCDH15; USH1G.
Conflict of interest statement
Ethics approval and consent to participate
The study was approved by the Institutional Review Boards of the Quaid-i-Azam University and the Baylor College of Medicine and Affiliated Hospitals (H-17566). Written informed consent was obtained from all participating members.
Competing interests
I.S. and W.A. are members of the BMC Medical Genetics editorial board. All other authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Yang T, Gurrola JG, Wu H, Chiu SM, Wangemann P, Snyder PM, et al. Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet. 2009;84:651–657. doi: 10.1016/j.ajhg.2009.04.014. - DOI - PMC - PubMed
-
- Mei L, Chen J, Zong L, Zhu Y, Liang C, Jones RO, et al. A deafness mechanism of digenic Cx26 ( GJB2 ) and Cx30 ( GJB6 ) mutations: reduction of endocochlear potential by impairment of heterogeneous gap junctional function in the cochlear lateral wall. Neurobiol Dis. 2017;108:195–203. doi: 10.1016/j.nbd.2017.08.002. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
