

Identifying Genes Whose Mutant Transcripts Cause Dominant Disease Traits by Potential Gain-of-Function Alleles
- PMID: 30032986
- PMCID: PMC6081281
- DOI: 10.1016/j.ajhg.2018.06.009
Identifying Genes Whose Mutant Transcripts Cause Dominant Disease Traits by Potential Gain-of-Function Alleles
Abstract
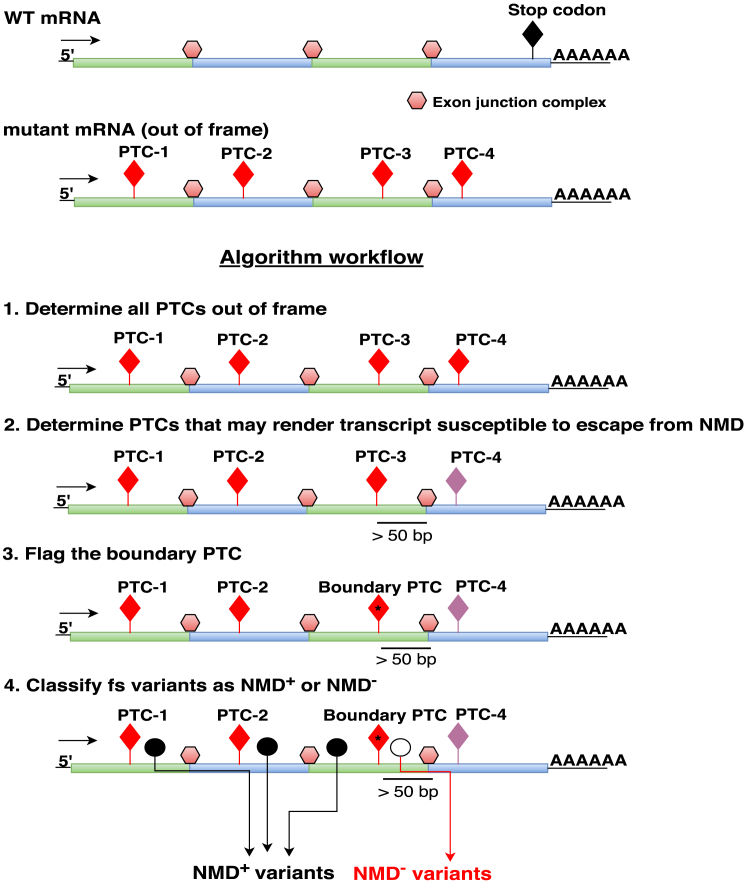
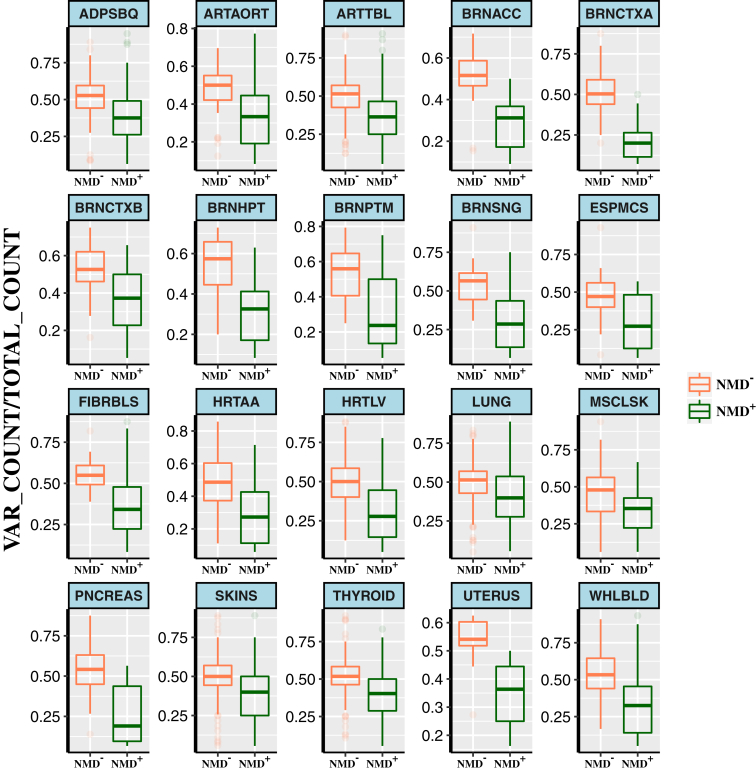
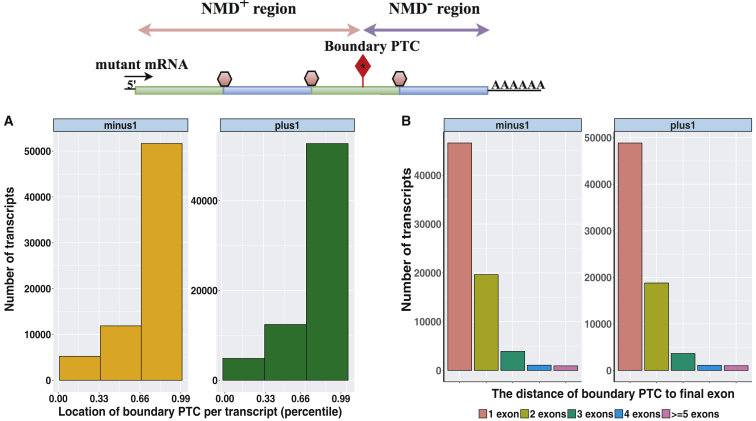
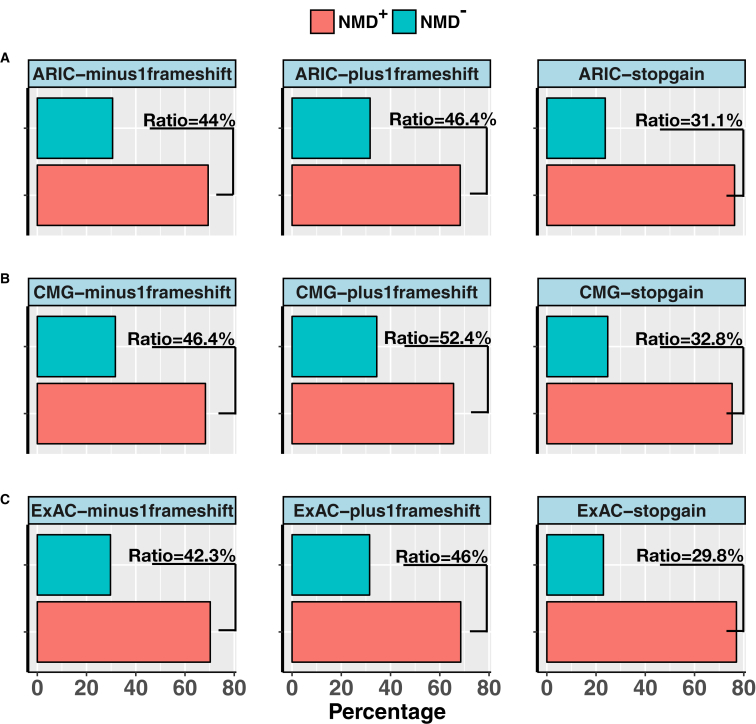
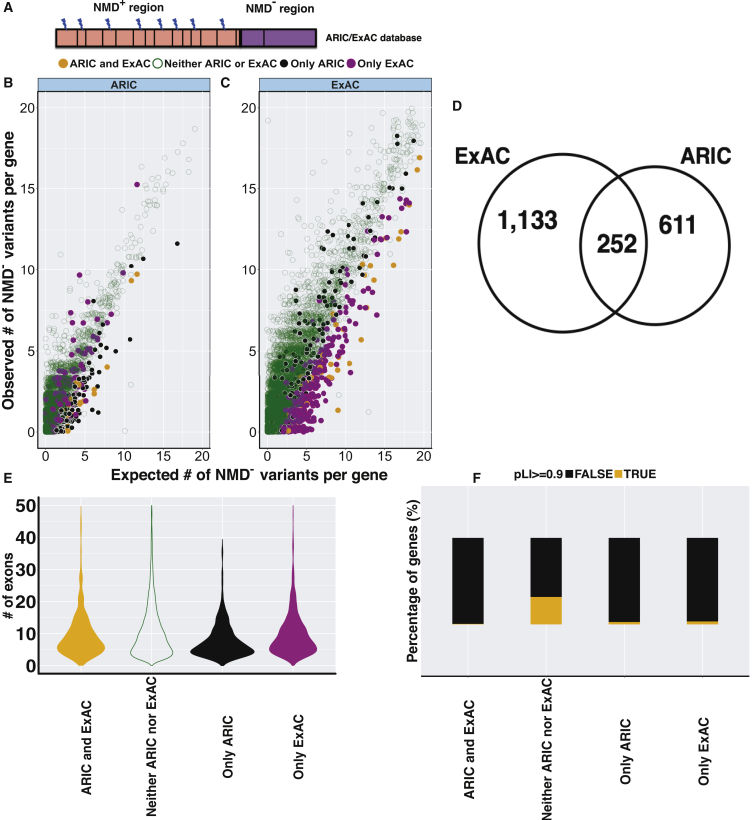
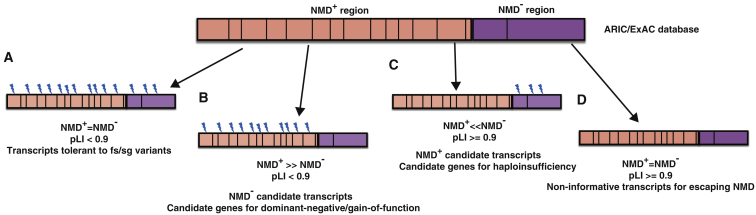
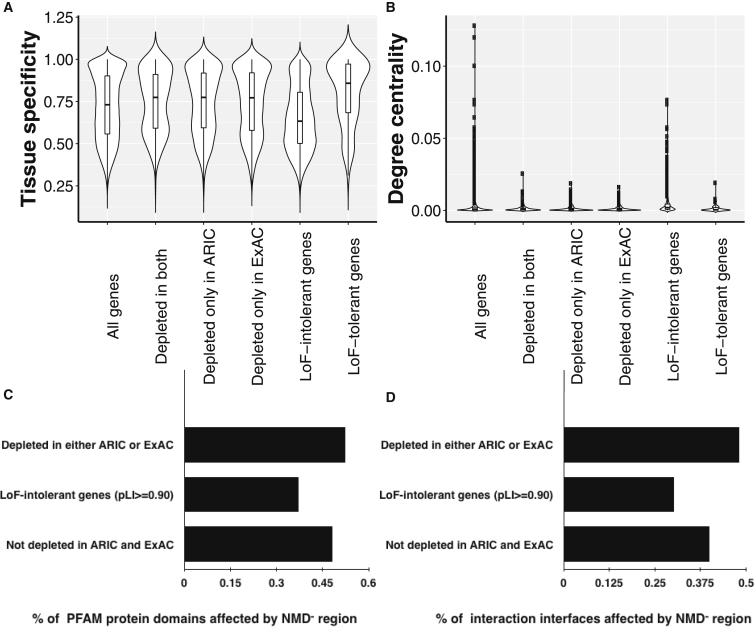
Premature termination codon (PTC)-bearing transcripts are often degraded by nonsense-mediated decay (NMD) resulting in loss-of-function (LoF) alleles. However, not all PTCs result in LoF mutations, i.e., some such transcripts escape NMD and are translated to truncated peptide products that result in disease due to gain-of-function (GoF) effects. Since the location of the PTC is a major factor determining transcript fate, we hypothesized that depletion of protein-truncating variants (PTVs) within the gene region predicted to escape NMD in control databases could provide a rank for genic susceptibility for disease through GoF versus LoF. We developed an NMD escape intolerance score to rank genes based on the depletion of PTVs that would render them able to escape NMD using the Atherosclerosis Risk in Communities Study (ARIC) and the Exome Aggregation Consortium (ExAC) control databases, which was further used to screen the Baylor-Center for Mendelian Genomics disease database. This analysis revealed 1,996 genes significantly depleted for PTVs that are predicted to escape from NMD, i.e., PTVesc; further studies provided evidence that revealed a subset as candidate genes underlying Mendelian phenotypes. Importantly, these genes have characteristically low pLI scores, which can cause them to be overlooked as candidates for dominant diseases. Collectively, we demonstrate that this NMD escape intolerance score is an effective and efficient tool for gene discovery in Mendelian diseases due to production of truncated or altered proteins. More importantly, we provide a complementary analytical tool to aid identification of genes associated with dominant traits through a mechanism distinct from LoF.
Keywords: NMD escape intolerance scores; NMDEscPredictor; WES analysis; antimorphic; bioinformatic tool; dominant negative; frameshift alleles; genotype-phenotype correlations; nonsense-mediated decay; stopgain variants.
Copyright © 2018 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Lykke-Andersen S., Jensen T.H. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015;16:665–677. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
